Синдром Александера (Alexander) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 21.01.2026
ГУЗ «Областная детская больница», Липецк, Россия
Младенческая форма болезни Александера (описание клинического наблюдения и обзор литературы)
Журнал: Журнал «Вопросы нейрохирургии» имени Н.Н. Бурденко. 2016;80(6): 93‑98
Представляем случай младенческой формы болезни Александера. Уникальность случая в том, что обследование пациентки началось на доклинической стадии и продолжено во время манифестации и в разгар болезни. Представлены редкие снимки, полученные при нейросонографии на доклиническом этапе заболевания, и уникальные результаты МРТ-исследований. Результаты МРТ-исследований в момент начала заболевания и спустя 3 года указывают на наличие клинических стадий в течении младенческой формы болезни Александера.
Болезнь Александера — редкое наследственное заболевание, сопровождающееся прогрессирующей дегенерацией белого вещества головного мозга. Причиной заболевания служит мутация гена, который кодирует белок GFAP (глиальный фибриллярный кислый белок). Russo и соавт. (1976) выделяют три формы заболевания в зависимости от времени манифестации: младенческая — от 0 до 2 лет, подростковая − от 2 до 12 лет и болезнь взрослых — старше 12 лет [1]. Некоторые авторы [2—4] описывают еще и неонатальную форму. От 63 до 80% случаев приходятся на младенческую форму [3, 5, 6].
В последнее время получила распространение классификация, опирающаяся на локализацию поражения и клиническую картину. Prust и соавт. (2011) выделяют I и II типы заболевания. К I типу относятся ранние формы с преимущественно фронтальной лейкодистрофией, ко II типу — преимущественно поражения мозжечка и стволовых структур. Медиана выживаемости для I типа составляет 14 лет, для II типа — 25 лет [1]. Существует мнение, что чем раньше манифестирует заболевание, тем более злокачественным является его течение. Это особенно касается неонатальной формы [2, 3]. В то же время некоторые специалисты отмечают непредсказуемость течения заболевания в любом возрасте и возрастную классификацию не считают полезной [2].
S. Marjo van der Knaap и соавт. [7] предлагают пять МРТ-критериев установления диагноза болезни Александера:
— обширные изменения белого вещества больших полушарий, преимущественно лобных долей;
— перивентрикулярный ободок с высоким сигналом в Т1- и низким уровнем сигнала в Т2-режимах;
— изменения базальных ганглиев и таламусов;
— поражение ствола мозга;
— повышение контрастности серого и белого вещества.
Четыре из 5 критериев достаточны для постановки диагноза [7].
У некоторых пациентов из-за сдавления сильвиева водопровода развивается гидроцефалия, в связи с чем эти больные требуют нейрохирургического вмешательства [1—3, 8].
Клиническое наблюдение
Представляем клинический случай девочки Т., которая впервые попала в поле зрения неврологов и нейрохирургов в возрасте 2 мес.
Ребенок от первой беременности, срочных родов в сроке 42 нед, протекавших самостоятельно, без осложнений. Масса тела при рождении — 3310 г, рост — 53 см. До 2 мес росла и развивалась без особенностей. В возрасте 2 мес была направлена на нейросонографию (НСГ) с целью профилактического осмотра. На момент проведения исследования мать не предъявляла жалоб, отмечая лишь незначительные эпизоды беспокойства ребенка.
Результат НСГ представлен на рис. 1.
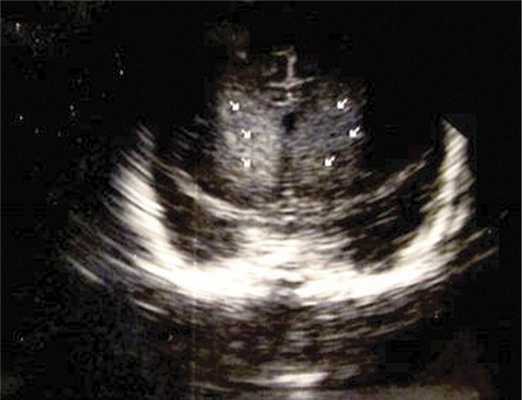
Рис. 1. Больная Т. НСГ в возрасте 2 мес.
Фронтальная стандартная проекция. Стрелками обозначены облаковидные гиперэхогенные зоны в перивентрикулярных отделах передних рогов боковых желудочков. Желудочковая система не расширена. Отмечается сужение передних рогов боковых желудочков в месте соприкосновения с областями повышенной эхогенности — результат объемного воздействия патологических очагов.
Учитывая необычность находки при НСГ, родителям было предложено стационарное обследование для уточнения диагноза. 28.06.11 ребенок поступил в отделение грудного возраста. Данные НСГ были расценены как возможное ишемическое поражение натального периода или объемный процесс. В отделении состояние катастрофически ухудшилось: перестала держать голову, появились хореоидные движения в лицевой мускулатуре и атетоидные движения в дистальных отделах конечностей, выраженные приступы беспокойства. 07.07.11 выполнена магнитно-резонансная томография (МРТ) головного мозга без контраста и с контрастным усилением Магневистом. Результаты представлены на рис. 2 и 3.
Рис. 2. Больная Т., возраст 2,5 мес. МРТ головы без контрастного усиления в Т1- и Т2-режимах. Определяется преимущественное поражение белого вещества лобных долей с вовлечением подкорковых ганглиев. Имеет место умеренное расширение боковых желудочков. Рис. 3. Больная Т., возраст 2,5 мес. МРТ головы с контрастным усилением. Симметричное накопление контраста в лобных долях, зрительных буграх и четверохолмии.
Очаговые изменения, выявленные при МРТ, в целом совпали с таковыми при выполнении НСГ. Хорошо заметен очаг в области четверохолмия. Уже на этих снимках можно констатировать появление вентрикуломегалии. Определяется объемное воздействие патологических очагов на передние рога боковых желудочков, просвет которых практически не определяется.
С 26.07.11 наступило дальнейшее ухудшение состояния в виде нарастания беспокойства, появления рвоты и приступов «замирания». По данным НСГ, отмечается прогрессирующее расширение желудочковой системы. 27.07.11 проведена операция вентрикулоперитонеостомии. Операция имела определенные особенности, продиктованные заболеванием: в связи с сужением передних рогов боковых желудочков, обусловленным объемным воздействием патологических очагов, принято решение провести вентрикулостомию не из точки Кохера, а через задний рог (точка Денди), проведя катетер за сосудистое сплетение с использованием ультразвукового контроля. В послеоперационном периоде отмечено улучшение состояния. Общемозговая симптоматика регрессировала, гиперкинезы уменьшились. Проведенный генетический анализ (генетическая лаборатория РДКБ, Москва) выявил мутацию c1117C >a (pClu373Lys) в гене GFAP (болезнь Александера), которая описана в базе данных по мутациям (cm023075) в гетерозиготном состоянии.
В дальнейшем ребенок наблюдался неврологом, получал антиконвульсантную терапию. Повторное комплексное обследование было проведено в возрасте 3 лет. Общее состояние ребенка расценено как удовлетворительное. Отмечается выраженная задержка психомоторного развития. Эпилептические приступы 1—2 раза в неделю в виде кратковременного «замирания». На момент осмотра в сознании, речь на уровне лепета, к осмотру позитивна, взгляд фиксирует, интересуется игрушками, берет их в руки, подолгу рассматривает, сидит без поддержки, не ходит. Расходящееся косоглазие. Хореоподобные движения в конечностях. Выраженная мышечная гипотония.
Выполнена МРТ без контраста и с контрастным усилением. Результаты представлены на рис. 4.
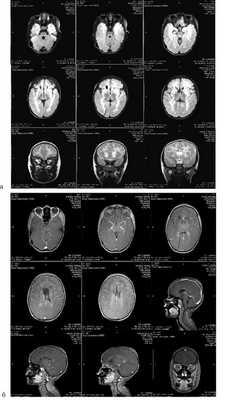
Рис. 4. Больная Т., возраст 3 года 2 мес. МРТ головы. Состояние после вентрикуло-перитонеального шунтирования (ВПШ). а — МРТ без контраста. Зоны поражения сохраняют прежнюю локализацию, но менее обширны. Нет признаков объемного воздействия патологических очагов. Просвет передних рогов хорошо прослеживается вместе с полостью прозрачной перегородки; б — МРТ головного мозга с контрастом. Очаги накопления контраста не определяются.
Описанный случай интересен в нескольких аспектах. С момента первого описания болезни в 1949 г. вплоть до 2011 г. было описано менее 100 случаев заболевания, и только около 50 — с использованием современных средств нейровизуализации. В 2011 г. С.В. Серков и соавт. [8] впервые в России представили два случая ювенильной формы заболевания. Наш случай, возможно, третий из выявленных в РФ.
Вторым немаловажным наблюдением являются данные, полученные при МРТ с контрастным усилением. Здесь важны характер накопления контраста и период болезни, в который это накопление происходит. На момент появления клинических симптомов зафиксировано интенсивное накопление контраста в очагах поражения. Наряду с интенсивностью накопления обращает на себя внимание симметричность очагов. Другая особенность — отсутствие накопления спустя 3 года после манифестации заболевания, в период стабилизации состояния. Можно предположить, что накопление контраста в очагах лейкодистрофии характерно для манифестации и разгара заболевания, а период ремиссии может характеризоваться отсутствием такового. В случае ювенильной формы болезни, описанной С.В. Серковым и соавт. [8], интенсивность накопления контраста не менялась. Описание обнаруженной в нашем случае особенности контрастирования в литературе нам не встречалось, возможно, это характерно для младенческой формы заболевания.
В период манифестации заболевания картина МРТ полностью соответствовала критериям, предложенным Marjo S. van der Knaap и соавт. [7], однако в период стабилизации клинических проявлений из описанных 5 критериев присутствовало не более 3.
Третий немаловажный факт касается самого течения младенческой формы заболевания. Болезнь может протекать от нескольких месяцев до 8 лет. Средняя продолжительность болезни составляет 2—3 года [8]. Наш случай демонстрирует длительный период стабилизации состояния. Можно предположить, что своевременно выполненная шунтирующая операция в иных случаях способствует стабилизации состояния пациента и продлению его жизни.
Анализ описанного наблюдения позволяет предполагать существование стадийности в течении младенческой формы болезни Александера.
Выводы
Выявленные при нейросонографии очаговые изменения в виде гиперэхогенных облаковидных очагов, распространяющихся из перивентрикулярных отделов передних рогов боковых желудочков на базальные ганглии, могут быть специфичными для младенческой формы болезни Александера. Очаги лейкодистрофии в период манифестации и разгара заболевания имеют вид объемного процесса. Очаги лейкодистрофии, определяемые при МРТ-иссле-довании, способны интенсивно накапливать контраст в период манифестации болезни, и, напротив, не накапливают контраста в период затихания клинических проявлений, что может свидетельствовать о наличии как минимум двух стадий болезни.
Младенческая форма болезни, возможно, не всегда быстро приводит к летальному исходу. В то же время тяжесть поражения белого вещества неотвратимо приводит к инвалидизации больного. Свое-временно проведенная шунтирующая операция, вероятно, способствует продлению жизни этих пациентов.
Конфликт интересов отсутствует .
Комментарий
Работа посвящена редкому генетическому заболеванию. Уникальным описанное наблюдение делают хорошая нейровизуализация стадий развития заболевания, редкие данные нейросонографии при наблюдаемой патологии. Также интересен обзор литературы, приведенный авторами в обсуждении. Безусловно, работа достойна публикации в нейрохирургическом журнале, поскольку расширяет кругозор нейрохирургов в вопросе дифференциальной диагностики. Кроме того, как показали авторы, представленное заболевание иногда требует и нейрохирургических вмешательств для облегчения состояния больного.
Синдром Александера (Alexander) - синонимы, авторы, клиника
Внимание! Обнаружена ошибка
К сожалению, данная страница для Вас недоступна, возможно был изменён её адрес или она была удалена. Пожалуйста, воспользуйтесь поиском.
Навигация
Популярное
Гипоаллергенная диета: меню и перечень продуктов
Список противоаллергических препаратов нового поколения
Перечень антигистаминных препаратов нового поколения
Благодаря нашим высококвалифицированным специалистам мы предоставляем высокое качество и эффективный результат.
Навигация
Контакты
Вы можете связаться с нами по любому нижеперечисленным контактам.
Болезнь Александера: симптомы, диагностика, лечение

Болезнь Александера (лейкодистрофия Розенталя, болезнь Розенталя) - это довольно редкое заболевание нервной системы, обусловленное генной мутацией. Симптомы развиваются в результате нарушения питания нервных клеток и блока проведения нервных импульсов по миелиновым волокнам. Клиническая картина болезни весьма разнообразна. Эффективных способов лечения пока не существует. В этой статье Вы сможете ознакомиться с причинами, симптомами, методами диагностики болезни Александера, а также возможностями оказания медицинской помощи таким больным.
Впервые заболевание было описано патологоанатомом Александером в 1949 году. Болезнь характеризуется неуклонно прогрессирующим течением. Распространенность болезни Александера достоверно не изучена ввиду малой встречаемости этой патологии. Описано около 500 случаев заболевания в США; проведенное в Японии исследование выявило наличие 1 случая заболевания на 2 700 000 человек. Теоретически подсчитана вероятность возникновения болезни Александера в человеческой популяции - 1:3 000 000. Точно известно, что возникновение заболевания не зависит от расы, пола, места проживания на Земном шаре.
Причины

В 95% случаев болезнь Александера развивается в результате мутации в гене, расположенном на 17-й хромосоме. Обычно мутация возникает спонтанно, то есть родители являются совершенно здоровыми, их генотип не имеет подобных изменений. Скорее всего, изменение гена происходит в отцовской хромосоме во время сперматогенеза, и если такой «аномальный» сперматозоид оплодотворяет яйцеклетку, то тогда и развивается болезнь у ребенка.
Ген отвечает за продукцию глиального фибриллярного кислого белка GFAP. В случае мутации измененный белок GFAP накапливается во вспомогательных клетках нейронов (нейроглии), что препятствует обеспечению нейронов питательными веществами. Кроме того, при болезни Александера в самом измененном белке GFAP образуются узелковые образования, которые называют волокнами Розенталя. Последние мешают нормальному проведению нервных импульсов по миелиновым волокнам.
У 5% людей, у которых диагностирована болезнь Александера, подобный или иной генетический дефект не обнаруживается, то есть причина развития остается неизвестной.
Симптомы
Заболевание впервые проявляет себя у людей в разном возрасте. В зависимости от этого принято выделять несколько клинических форм:
- инфантильная (младенческая);
- ювенильная (юношеская);
- взрослая.
Предполагается наличие так называемой неонатальной формы заболевания, когда ребенок рождается уже с проявлениями болезни. У таких детей обычно с первых дней жизни отмечается повышенное внутричерепное давление, аномально большой череп. Характерен судорожный синдром, выраженная задержка нервно-психического развития. К сожалению, продолжительность жизни таких деток не составляет и года. Некоторые ученые относят эту форму к инфантильной, но с очень ранним началом.
Инфантильная форма развивается в раннем детском возрасте, в среднем - по достижении 6 месяцев. У таких детей плохой аппетит, они часто срыгивают вплоть до рвоты. Отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления. Естественно, что это сказывается на темпах физического и нервно-психического развития. Дети плохо прибавляют в весе, поздно начинают держать голову (после 3-х месяцев), садиться и ползать. По мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений. На фоне парезов в конечностях появляются непроизвольные движения: выкручивающие, червеобразные движения в пальцах рук, повороты головой с фиксацией позы и тому подобное. Эти явления называются гиперкинезами, в частности, хореоатетозом. Возможны судорожные эпилептические припадки. Страдает интеллект: дети не узнают близких, их не радуют игрушки, они не овладевают навыками (например, не могут нанизать кольца на пирамидку в соответствующем возрасте). Также нарушается координация движений, наблюдаются подергивания глазных яблок (нистагм). Самостоятельная ходьба практически невозможна. Заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма проявляет себя несколько позже, в возрасте от 4 до 14 лет, в среднем - около 9 лет. Хотя отдельные признаки заболевания могут появиться и раньше - в 2-3 года, но обычно их не связывают с болезнью Александера. Такие детки несколько отстают в нервно-психическом развитии, страдают от судорог. У них голова имеет больший размер по сравнению со сверстниками (но не настолько больший по сравнению с инфантильной формой). Несколько позже присоединяются нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды. Голос приобретает гнусавый оттенок. Движения языком затрудняются. Все эти изменения формируют бульбарные и псевдобульбарные расстройства, а возникают в результате поражения ствола мозга. По утрам больных беспокоят неукротимые рвоты. Так же, как и при инфантильной форме, появляется мышечная слабость в конечностях, которая постепенно нарастает.
Мышечный тонус увеличивается, мышцы становятся плотными и твердыми на ощупь, появляются патологические стопные признаки (симптом Бабинского и другие). Постепенно эти изменения охватывают все четыре конечности, что становится причиной расстройств передвижения и самообслуживания. Возможны нарушение равновесия, расстройства поведения. Обычно интеллектуальные расстройства выражены незначительно или вообще отсутствуют, хотя описаны случаи и резкого снижения мыслительных способностей. У больных с ювенильной формой периодически регистрируют рефлекторную остановку дыхания: апноэ. В конце концов, прогрессирующее поражение нервной системы заканчивается смертельным исходом, в среднем, через 10 лет от появления начальных клинических признаков заболевания.
Взрослая форма развивается в сроки от 20 до 70 лет. Клинические симптомы довольно разнообразны, поскольку могут быть отражением патологии любого участка головного мозга. Чаще всего это парезы и параличи с повышенным мышечным тонусом, нарушения координации движений и равновесия, непроизвольные неконтролируемые движения, нарушения речи и глотания. Снижение интеллекта незначительное. Часто выявляется нистагм и нарушение содружественных (одновременных и однонаправленных) движений глазными яблоками. Болезнь прогрессирует и неизбежно заканчивается летальным исходом (обычно от присоединения интекуррентных инфекций).
Диагностика
Диагностика заболевания прижизненно довольно затруднительна, потому что клинических симптомов, свойственных только болезни Александера, нет. И специфических изменений ни один из методов исследования не выявляет (не считая генетического анализа, который, тем не менее, необходимо еще назначить, подозревая эту болезнь).
При магнитно-резонансной томографии головного мозга (МРТ) при болезни Александера выявляется демиелинизация различных отделов мозга (при инфантильной и юношеской формах — преимущественно в лобных с распространением на другие области, при взрослой - более выражена в мозжечке и стволе мозга).
При электроэнцефалографии регистрируют изменения биоэлектрической активности мозга в лобных отделах.
Генетический анализ наиболее точно позволяет подтвердить диагноз болезни Александера: находят мутацию в гене GFAP на 17-й хромосоме (в 95% случаев). Следует помнить, что у 5% больных этим заболеванием генетический дефект не обнаружен и по сей день.
Подтверждением заболевания служит обнаружение волокон Розенталя (что возможно при биопсии мозга или уже после смерти при вскрытии).
Лечение
Сегодня медицина не располагает эффективными способами лечения болезни Александера. Возможно, будущее в этом направлении принадлежит генной инженерии.
После установления диагноза обычно проводят симптоматическую терапию, позволяющую облегчить и продлить жизнь больному:
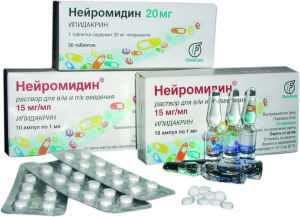
- при парезах назначают стимуляторы нервно-мышечной проводимости (Нейромидин);
- при спастичности мышц - миорелаксанты (Баклофен, Сирдалуд, Мидокалм);
- при эпилептических припадках - противосудорожные препараты (Вальпроаты, Сибазон и другие);
- для уменьшения непроизвольных движений могут использоваться нейролептики (Галоперидол, Азалептин и другие).
Для передвижения используют специальные приспособления, в том числе и ортопедические. Пик болезни позволяет передвигаться только с помощью инвалидной коляски. Конечно, в терминальных стадиях заболевания больные нуждаются в постоянном постороннем уходе.
Болезнь Александера - редкое, в основном, генетически обусловленное заболевание. Его развернутая клиническая картина представляет собой двигательные, координаторные нарушения, проблемы с речью и приемом пищи. Почти все взрослые больные живут не более 10 лет с момента развития заболевания. Наиболее точным методом диагностики является генетический. Способы лечения находятся в стадии разработки, больным в настоящее время помогают только симптоматическими средствами.
Болезнь Александера
Болезнь Александера является одним из группы неврологических заболеваний, известных как лейкодистрофии. Лейкодистрофии — это расстройства, возникающие в результате нарушений в миелине, «белом веществе», которое защищает нервные волокна в мозге. При болезни Александера разрушение белого вещества сопровождается образованием розентальных волокон — аномальных скоплений белка, которые накапливаются в ненервных клетках (астроцитах) в мозге.
Наиболее распространенным типом болезни Александра является инфантильная форма, которая обычно начинается в течение первых двух лет жизни ребенка. Симптомы включают умственные и физические задержки в развитии, сопровождаемые потерей основных этапов развития, ненормальным увеличением размера головы и судорогами (эпилепсия). Ювенильная форма болезни Александера начинается в возрасте от двух до тринадцати лет. У этих детей может быть чрезмерная рвота, трудности с глотанием и речью, плохая координация и потеря контроля над двигательными функциями. Взрослые формы болезни Александера встречаются реже. Симптомы иногда имитируют симптомы болезни Паркинсона или рассеянного склероза или могут проявляться главным образом как психическое расстройство.
Заболевание встречается как у мужчин, так и у женщин, и в его распространении нет этнических, расовых, географических или культурных/экономических различий. Болезнь Александера является прогрессирующим и часто смертельным заболеванием.
Причины болезни Александера
Около 95% случаев болезни Александера вызваны мутациями в гене, называемом GFAP, в отношении структурного белка, называемого глиальным фибриллярным кислым белком, который обнаруживается исключительно в астроцитах в центральной нервной системе (ЦНС). Причина других 5% случаев неизвестна.
Мутации GFAP доминируют. Доминантные генетические нарушения возникают тогда, когда для того, чтобы вызвать ту или иную болезнь, требуется лишь одна копия аномального гена. Таким образом, у пациентов с заболеванием Александера есть одна копия мутированного и одна нормальная копия гена GFAP. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения в ДНК гена).
Как мутации GFAP приводят к болезни Александра, неизвестно.
Симптомы болезни Александера
Расстройство впервые проявляет себя у людей в разном возрасте. В зависимости от этого принято выделять несколько клинических форм:
Инфантильная форма развивается в раннем детском возрасте, в среднем — по достижении 6 месяцев. У таких детей плохой аппетит, они часто срыгивают вплоть до рвоты. Отмечается патологически быстрое увеличение размеров головы, нарастание внутричерепного давления. Естественно, что это сказывается на темпах физического и нервно-психического развития. Дети плохо прибавляют в весе, поздно начинают держать голову (после 3-х месяцев), садиться и ползать.
По мере роста и развития ребенка развивается мышечная слабость в конечностях (парезы) наряду с повышенным мышечным тонусом (спастичность), что проявляется ограничением объема и силы произвольных движений. На фоне парезов в конечностях появляются непроизвольные движения: выкручивающие, червеобразные движения в пальцах рук, повороты головой с фиксацией позы и тому подобное. Эти явления называются гиперкинезами, в частности, хореоатетозом. Возможны судорожные эпилептические припадки. Страдает интеллект: дети не узнают близких, их не радуют игрушки, они не овладевают навыками (например, не могут нанизать кольца на пирамидку в соответствующем возрасте). Также нарушается координация движений, наблюдаются подергивания глазных яблок (нистагм). Самостоятельная ходьба практически невозможна. Заболевание неуклонно прогрессирует и заканчивается смертью в течение 2-3 лет.
Ювенильная форма проявляет себя несколько позже, в возрасте от 4 до 14 лет, в среднем — около 9 лет. Хотя отдельные признаки заболевания могут появиться и раньше — в 2-3 года, но обычно их не связывают с болезнью Александера. Такие детки несколько отстают в нервно-психическом развитии, страдают от судорог. У них голова имеет больший размер по сравнению со сверстниками (но не настолько больший по сравнению с инфантильной формой). Несколько позже присоединяются нарушения речи (смазанность, нечеткость), поперхивание при приеме пищи, а затем и при глотании воды. Голос приобретает гнусавый оттенок. Движения языком затрудняются. Все эти изменения формируют бульбарные и псевдобульбарные расстройства, а возникают в результате поражения ствола мозга. По утрам больных беспокоят неукротимые рвоты. Так же, как и при инфантильной форме, появляется мышечная слабость в конечностях, которая постепенно нарастает.
Лечение болезни Александера
- при парезах назначают стимуляторы нервно-мышечной проводимости (Нейромидин);
- при спастичности мышц - миорелаксанты (Баклофен, Сирдалуд, Мидокалм);
- при эпилептических припадках - противосудорожные препараты (Вальпроаты, Сибазон и другие);
- для уменьшения непроизвольных движений могут использоваться нейролептики (Галоперидол, Азалептин и другие).
Прогноз
Прогноз для людей с болезнью Александера, как правило, плохой. Большинство детей с инфантильной формой живут дольше 6 лет. Юношеские и взрослые формы расстройства протекают медленнее и продолжительнее.

Врач-терапевт, гастроэнтеролог, гепатолог, инфекционист. Провожу профилактические мероприятия осложнений со стороны пищеварительной системы после долгой терапии НПВП и кроворазжижающими препаратами.

Болезнь Александера - это довольно редкое заболевание нервной системы, обусловленное генной мутацией. Симптомы развиваются в результате нарушения питания нервных клеток и блока проведения нервных импульсов по миелиновым волокнам.
Причины
Клиническая картина
Диагностика
Лечение
Читайте также: