Синдром Арнольда-Киари (Arnold-Chiari) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 28.01.2026
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14-30 % популяции
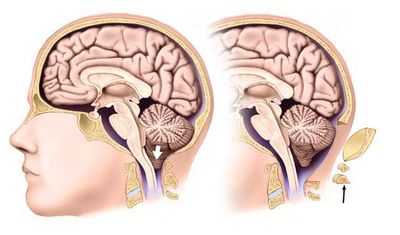
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4-6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835-1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851-1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835-1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Гидроцефалия — патологическое состояние, характеризующееся избыточным накоплением спинномозговой жидкости (ликвора) в ликворных пространствах головного мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891-1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15-20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
Менингомиелоцеле — грыжа спинномозгового канала, при которой происходит выпячивание тканей и вещества спинного мозга через костный дефект позвоночного столба
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20-40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2-3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50-85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Аномалия Киари ( Синдром Арнольда-Киари )
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
МКБ-10

Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
- Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
- Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
- Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
- Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Валери Торро. Тракционная миелопатия, Синдром Арнольда-Киари I, Дископатия

Я бы хотела дополнить свое свидетельство спустя 10 лет после рассечения концевой нити в Барселоне.
Я была прооперирована в 2009 году доктором Ройо. Могу сказать, что хорошо себя чувствую, даже очень хорошо, если сравнивать с состоянием в 2009 году. Большинство симптомов исчезло, головные боли сейчас незначительные. Если они появляются, то слабо, и быстро проходят, в большинстве случаев я могу их выдержать без лекарств.
К сожалению, я вернулась в Барселону, но не из-за себя, а из-за моей дочери, которой тоже поставили синдром Арнольда Киари.
Конечно, во Франции врачи говорят, что это не наследственное заболевание. Однако у дочери такая же патология, как у меня, странно, правда? Несмотря на то, что это заболевание не является генетическим для французского здравоохранения.
Мы без всяких сомнений поехали в Барселону.
Надеюсь, что для нее операция тоже станет успешной, чтобы она могла спокойно жить дальше.
Я надеюсь, это свидетельство поможет тем пациентам, что сомневаются.
Я бы хотела поблагодарить команду Барселоны за гостеприимство и, особенно, за их знания, которые помогают больным жить полной жизнью.
Дата операции: февраль 2009
Меня зовут Валери, я живу во Франции, мне 43 года и я страдаю Синдромом Арнольда-Киари первого типа.
Все началось после моих первых родов 11 лет назад. У меня начались сильные головные боли, когда я смеялась, кашляла или чихала. Со временем боли участились и стали практически постоянными, очень сильными и неконтролируемыми.
Я начала искать в Интернете, пока не нашла сайт доктора Ройо. Я сразу же сконтактировала с некоторыми из прооперированных им французских пациентов, и узнала их истории.
Два года назад, в 2007, я больше не могла терпеть эти боли и попросила своего врача сделать мне МРТ для контроля протрузий межпозвоночных дисков, которые у меня есть. И тогда-то открылся диагноз: Синдром Арнольда-Киари. Я обратилась к неврологу и нейрохирургу, так как боли стали невыносимыми, и я не могла работать.
Нейрохирург мне сказал, что меня можно было прооперировать. Но, пока возможно терпеть боль, лучше ничего не делать, так как нельзя гарантировать, что эти боли исчезнут. Я решила ничего не делать, так мысль о том, чтобы открыть череп и, возможно, безрезультатно, меня приводила в шок.
В июне 2008 года мое состояние очень стремительно ухудшилось, за 6 месяцев появились новые симптомы:
- Потеря равновесия;
- Потеря силы в ногах и руках;
- Осложненное глотание;
- Утяжеленное дыхание;
- Боль в ребрах и груди;
- Постоянная астения;
- Парестез в руках и ногах;
- Недержание;
- Окаменелость в шее из-за сильных болей при движении;
- Невозможность находится долгое время сидя без движения;
- И, прежде всего, головные боли, которые только проходили лежа на спине, и общее необъяснимое недомогание.
Я приняла решение пойти на прием к доктору Ройо в феврале 2009 года, потому что не хотела, чтобы мне сделали краниектомию, традиционно практикуемую во Франции операцию. Я пришла на прием в понедельник, была прооперирована во вторник и выписана в среду. Какова была моя радость, когда я поняла, что голова больше не болит! Я смогла вернуться во Францию в сидячем положении, в то время, как на пути туда я ехала лежа.
Сейчас прошли 7 месяцев с дня операции, и я чувствую себя гораздо лучше. Несмотря на то, что некоторые симптомы остались, я должна отметить следующее:
- Головных болей почти нет, они уже не устанавливаются надолго, как раньше, и проходят быстро и без лекарств.
- Нет хронической усталости, хотя могу быть просто уставшей в конце дня, как все.
- Прошли боли в ребрах и груди.
- Нет затрудненного дыхания.
- Более свободные движения головой.
- Полностью прошло недержание.
Иногда все еще теряю равновесие, когда произвожу слишком резкие движения, но парестезия проявляется все меньше. Надо иметь терпение, не стоит торопить события, потому что время сделает все остальное, а у моих дочек снова появилась мама, которая улыбается.
Благодарю доктора Ройо и его коллектив за то, что они вернули мне желание жить.
Свяжитесь с нами

Меня зовут Нина, я буду ассистентом в Вашей консультации.
Все консультации, полученные через этот формуляр или по электронной почте Барселонского Института Киари & Сирингомиелии & Сколиоза, передаются медицинскому отделу, и ответы проверяются доктором М. Б. Ройо Сальвадор.
Синдром Арнольда Киари (мальформация или аномалия Арнольда Киари)
Последнее редактирование: 11/02/2019, Доктор Мигель Б. Ройо Сальвадор, Номер в коллегии: 10389. Нейрохирурга и Невролога.
Определение
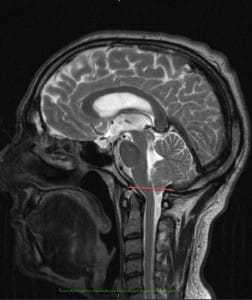
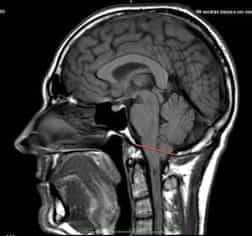
Синдром Арнольда Киари I типа состоит в опущении нижней части мозжечка - миндалин мозжечка - в затылочное отверстие и позвоночный канал при отсутствии других мальформаций, связанных со спинным мозгом. Иногда считают, что опущение миндалин мозжечка должно быть более 5мм, иногда - больше 3мм, для других опущение начинается с 0мм, когда миндалины находятся на уровне границы затылочного отверстия, при наличии соответствующей клинической картины.
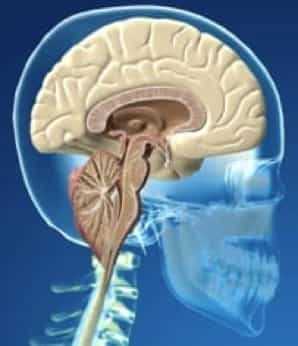
Рисунок 1.- На схеме показано опущение миндалин мозжечка и увеличение надмозжечкового пространства из-за смещения всего мозжечка к затылочному отверстию. Синдром Арнольда Киари I.
Симптомы
Клиническая картина при синдроме Арнольда Киари I может быть разнообразной, наиболее частые симптомы (от более частых к более редким): головные боли, боли в шейном отделе позвоночника, парез в конечностях, нарушения зрения, боль в конечностях, парестезии, нарушения чувствительности, головокружения, нарушения глотания, боли в пояснице, ухудшение памяти, нарушение походки, боли в грудном отделе позвоночника, нарушение равновесия, дисестезия, трудности с выражением мысли и подбором слов, нарушения сфинктера, бессонница, рвота, потери сознания, дрожь.
Типы Арнольда Киари
Существует 4 классических типа (I, II, III, IV) и два типа, описанных недавно (“0”, “1,5”):
I тип. Опущение миндалин мозжечка без какой-либо иной мальформации нервной системы.
II тип. Опущение миндалин мозжечка с нейропозвоночной мальформацией, при которой спинной мозг прикреплен к позвоночному каналу.
III тип. Опущение миндалин мозжечка с затылочной энцефалоцеле и мозговыми аномалиями при Синдроме Арнольда Киари II.
IV тип. Опущение миндалин мозжечка, аплазия или гипоплазия мозжечка, связанные с аплазией намёта мозжечка.
“1,5” тип. Недавно описан Синдром Арнольда Киари 1,5 с опущением миндалин мозжечка и опущением ствола головного мозга в затылочное отверстие.
Теории о причине опущения миндалин мозжечка
Опущение миндалин мозжечка может быть результатом натяжения, которое оказывает на спинной мозг та или иная мальформация, за исключением синдрома Арнольда Киари I типа, при котором опущение миндалин мозжечка - единственное нарушение, и о причине появления этого нарушения существуют разные теории:
- Традиционные теории:
- Гидродинамическая: опущение миндалин мозжечка - это последствие нарушения циркуляции спинно-мозговой жидкости.
- Мальформация: Теория о маленьком размере затылочного отверстия, которое провоцирует опущение миндалин в позвоночный канал.
- Теория натяжения спинного мозга по Filum System ® :
Теория доктора Мигеля Б. Ройо Сальвадор рассматривает опущение миндалин мозжечка при Синдрома Арнольда Киари I, как результат анормального натяжения спинного мозга из-за анормальной и натянутой связки, которая называется концевая нить. Концевая нить не видна на снимках.
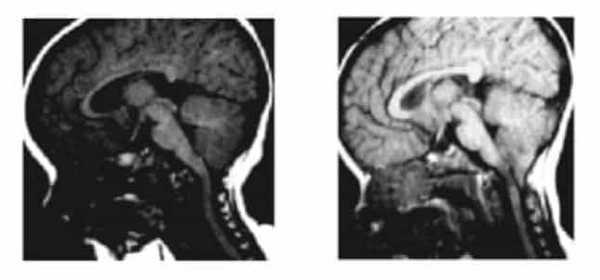
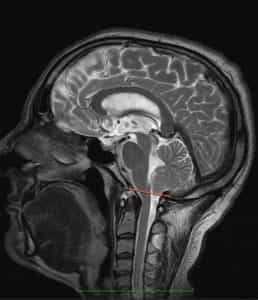
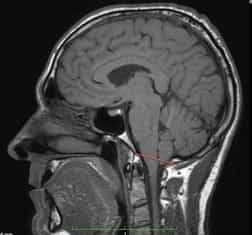
Рисунок 2.- МРТ пациента в 8 и 20 месяцев, на втором снимке можно наблюдать опущение миндалин мозжечка, которое появилось уже после первого МРТ. Huang P. “Adquired” Chiari I malformation. J. Neurosurg 1994. Это указывает на то, что, помимо наследственного и генетического фактора, существует фактор приобретенного заболевания.
Факторы, влияющие на развитие Синдрома Арнольда Киари I
Основные факторы, участвующие в развитии Синдрома Арнольда Киари I, следующие:
- Семейная предрасположенность: Натяжение нервной системы из-за анормально напряженной концевой нити, которое мы называем заболеванием концевой нити, приводит к опущению миндалин мозжечка. Эта патология является генетической и может передаваться по наследству.
- Внезапное усиление натяжения спинного мозга: После падения или травмы позвоночника уже существующее натяжение спинного мозга - заболевание концевой нити - может усилиться. В этих случаях из-за более сильного натяжения может увеличиться опущение миндалин мозжечка или компрессия в затылочном отверстии, что отразится на росте симптомов. Травма, в данном случае, не является причиной развития синдрома Арнольда Киари, это лишь пусковой механизм для внезапного ухудшения состояния из-за увеличения силы натяжения.
Осложнения при Синдроме Арнольда Киари I
Осложнения при пролабировании миндалин мозжечка могут зависеть от степени натяжения или от компрессии тканей в затылочном отделе (компрессия тканей миндалин мозжечка и ствола спинного мозга из-за ограниченности пространства в затылочном пространстве).
- Ухудшение качества жизни: при синдроме Арнольда Киари I головные боли, головокружения, боли в спине и конечностях, парезия, нарушения глотания или чувствительности, когнитивные нарушения, нарушения зрения или походки могут стать хроническими, увеличивать интенсивность, ухудшая с каждым разом состояние больного, ограничивая его нормальный образ жизни.
- Хронические боли: Пациентам может понадобиться лечение в отделении терапии боли, потому что привычные противовоспалительные и обезболивающие медикаменты могут быть неэффективными для лечения приступов головной боли и других симптомов.
- Внезапная смерть: в стволе спинного мозга находится контроль за сердечно-дыхательными функциями, если миндалины мозжечка давят на него, то во время сна могут возникать нарушения дыхания (апноэ, остановки дыхания и даже внезапная смерть пациента). Поэтому так важен своевременный диагноз и раннее лечение.
Лечение Синдрома Арнольда Киари I
Обычно при Синдроме Арнольда Киари I с/без сирингомиелии применяется нейрохирургическое лечение.
В настоящее время декомпрессия или краниотомия затылочного отверстия является стандартным лечением для этого диагноза в большинстве медицинских центров в мире. Обычно ее назначают в случаях, когда симптомы провоцируют больше ущерба и смертности, чем естественное развитие патологии.
Однако с 1993 года, после публикации докторской диссертации доктора Ройо Сальвадор, который связал натяжение всей нервной системы с концевой нитью, натяжение которой провоцирует, среди прочих заболеваний, опущение миндалин мозжечка, было разработано новое лечение, этиологическое, то есть, устраняющее причину заболевания при помощи хирургического рассечения концевой нити, которое убирает патологический механизм натяжения.
Наша техника рассечения концевой нити является минимально инвазивной, и ее назначают во всех случаях заболевания концевой нити, как симптоматического, так и протекающего без симптомов, и как можно раньше, поскольку риски минимальны и намного ниже, чем от самой патологии, к тому же, данное лечение останавливает дальнейшее развитие заболевания.
Рассечение концевой нити с техникой Filum System®:
Преимущества
1. Устраняет причину Синдрома Арнольда Киари I и связанных с ним патологий.
2. Устраняет давление в затылочном отверстии и вместе с ним риск внезапной смерти.
3. 0% смертности, без последствий у более чем 1500 прооперированных по методу Filum System® пациентов.
4. С минимально инвазивной техникой, хирургическое время: 45 минут. Короткие сроки пребывания в клинике. Местная анестезия. Короткий период постоперационного восстановления без ограничений.
5. Улучшает симптомы, останавливает опущение миндалин мозжечка.
6. Устраняет риск появления гидроцефалии из-за опущения миндалин.
7. Улучшает кровоснабжение всей нервной системы и, вместе с этим, когнитивные возможности, которые могут страдать из-за натяжения спинного мозга.
Недостатки
1. Небольшой шов в зоне копчика, возможны осложнения в виде инфекции шва и гематомы в зоне операции.
2. Улучшение спастичности иногда ошибочно принимают за снижение силы в конечностях.
3.Во время процесса восстановления и улучшения чувствительности могут появиться неприятные ощущения, которые обычно принимают за нежелательные последствия.
4. При улучшении кровоснабжения головного мозга может увеличиться мозговая активность и могут наблюдаться перепады в настроении в течение начального пост-оперативного периода.
Затылочная краниоктомия:
(Декомпрессия большого затылочного отверстия)
1. Избежание риска внезапной смерти.
2. Состояние некоторых пациентов улучшается.
1. Не устраняет причину заболевания.
2. Смертность от 0,7 до 12%, бóльший процент, чем внезапная смерть при спонтанном развитии заболевания.
3. Агрессивная операция, калечит и возможны последствия.
4. Малый процент улучшения и на короткий срок.
5. Неврологический дефицит: зависит от местонахождения увечья: Эмипарезия (паралич половины тела от 0,5 до 2,1%. Изменения в зрительном пространстве от 0,2 до 1,4%. Изменения в речи от 0,4 до 1%. Недостаток в чувствительности от 0,3 до 1%. Отстутствие равновесия, трудности с ходьбой от 10 до 30%.
6. Послеоперационное внутримозговое кровоизлияние в оперируемой области, эпидуральная гематома, интрааксиальное кровоизлияние, которые могут вызвать неврологический дефицит или ухудшение ранее существовашего дефицита (от 0,1 до 5%).
7. Отек мозга, в зависимости от процесса и ситуации, риск достигает 5%.
8. Поверхностная, глубокая или внутричерепная инфекция, риск от 0,1 до 6,8%, с формированием мозгового абсцесса, асептического-септического менингита.
9. Гемодинамические изменения из-за манипуляций с нарушениями в стволе мозга.
10. Газовая эмболия (у больных в сидячей позиции).
11. Выход спинномозговой жидкости от 3 до 14% (фистула СПЖ).
12. Послеоперационная гидроцефалия.
14. Тетрапарез (потеря силы во всех конечностях)
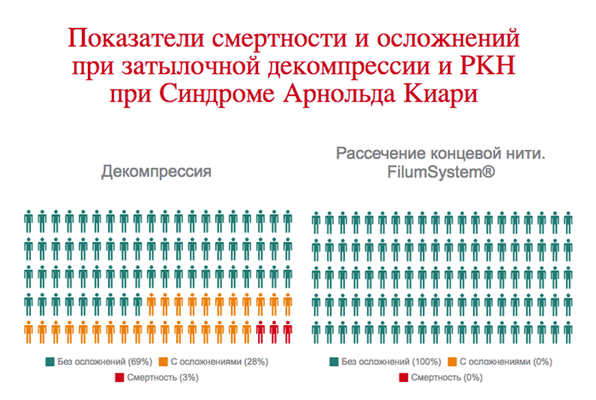

В связи с осложнениями и смертностью при затылочной декомпрессии, которые в процентном соотношении превышают смертность от спонтанного развития синдрома Арнольда Киари I, мы считаем, что данное лечение противопоказано.
Результаты лечения по методу Filum System
С помощью метода Filum System® уже прооперировалось более 1500 пациентов с Синдромом Арнольда Киари I, с/без сирингомиелией и/или сколиозом.
Цель операции: остановить развитие заболевания (как анатомическое (увеличение опущения миндалин), так и симптоматическое (дальнейшее развитие симптомов)). В большинстве случаев отмечается симптоматическое улучшение состояния, и в некоторых случаях миндалины мозжечка поднимаются.
BIBLIOGRAFÍA
- Dr. Miguel B. Royo Salvador (1996), Siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas, etiología común (PDF). REV NEUROL (Barc); 24 (132): 937-959.
- Dr. Miguel B. Royo Salvador (1996), Platibasia, impresión basilar, retroceso odontoideo y kinking del tronco cerebral, etiología común con la siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas (PDF). REV NEUROL (Barc); 24 (134): 1241-1250
- Dr. Miguel B. Royo Salvador (1997), Nuevo tratamiento quirúrgico para la siringomielia, la escoliosis, la malformación de Arnold-Chiari, el kinking del tronco cerebral, el retroceso odontoideo, la impresión basilar y la platibasia idiopáticas (PDF). REV NEUROL; 25 (140): 523-530
- M. B. Royo-Salvador, J. Solé-Llenas, J. M. Doménech, and R. González-Adrio, (2005) “Results of the section of the filum terminale in 20 patients with syringomyelia, scoliosis and Chiari malformation“.(PDF). Acta Neurochir (Wien) 147: 515-523.
- M. B. Royo-Salvador (2014), “Filum System ® Bibliography” (PDF).
- M. B. Royo-Salvador (2014), “Краткое Введение в Filum System ® “.
Синдром Арнольда Киари I
Описан: В 1883 году хирургом-анатомом Джоном Клеланд (1835-1925) из Пертшира, Шотландия. Он описал удлинение червя мозжечка, опущение мозжечка и IV желудочка у мальчика с гидроцефалией, энцефалоселе, спина бифида. В 1891 и 1896 годах Ганс Киари описал новые случаи и классифицировал их. В 1894 году Жюль Арнольд внес вклад в распространение знаний об этом заболевании.
Назван: Швальбе и Гредиг в 1907 году, как “мальформация Арнольда-Киари”. Официальная современная номенклатура в кодификации и классификации заболеваний Всемирной организации здравоохранения называет это заболевание термином “Синдром или заболевание Арнольда-Киари I” (Q07.0, CIE-10). (ВОЗ, (International Statistical Classification of Diseases and Related Health Problems, 10th Revision (c) Geneva, OMS, 1992)).
Частота: Один случай на тысячу новорожденных, другие авторы указывают менее 1% населения. В обоих случаях речь идет об опущении миндалин мозжечка более 3 или 5мм.
Заболевание концевой нити
После исследований доктора Ройо Сальвадор и его докторской диссертации (1992г.) было установлено, что несколько заболеваний, чья причина появления ранее была неизвестна, такие как: синдром Арнольда Киари I, идиопатические Сирингомиелия и Сколиоз, Платибазия, Базиллярная Импрессия, Смещение зуба осевого позвонка, Углообразный перегиб на уровне дуги атланта, - входят в состав новой патологии - Заболевания концевой нити - и возникают по одной и той же причине: натяжения спинного мозга и всей нервной системы.
Сила натяжения всей нервной системы при заболевании концевой нити присутствует при формировании всех человеческих эмбрионов, в большей или меньшей степени все страдают от ее последствий и разных форм проявлений и интенсивности.
С заболеванием концевой нити могут быть связаны такие заболевания как: межпозвонковые грыжи, некоторые синдромы сосудистой недостаточности головного мозга, фасеточный синдром, синдром Бострупа, фибромиалгия, хроническая усталость, ночной энурез, недержание мочеиспускания и острый парапарез.
Для точной диагностики, подбора лечения и наблюдения пациента с заболеванием концевой нити был создан метод Filum System ® .
Аномалия Арнольда-Киари: причины, синдром и симптомы, диагноз, лечение
Аномалию Арнольда-Киари относят к порокам кранио-вертебральной зоны. Формируется она в задней черепной ямке (ЗЧЯ), при недостаточном объеме которой происходит смещение задних отделов мозга и мозжечка в сторону большого затылочного отверстия, а также нарушается ток спинномозговой жидкости.
Задняя часть черепа образует так называемую заднюю черепную ямку, в которой расположены полушария и червь мозжечка, мост, продолговатый мозг, переходящий в спинной после прохождения сквозь большое затылочное отверстие. Большое затылочное отверстие ограничено костной основой черепа и не способно менять диаметр, любые смещения структур мозга чреваты несоответствием их размеров и диаметра отверстия, вклиниванием и ущемлением нервной ткани, последствия которого могут стать фатальными.
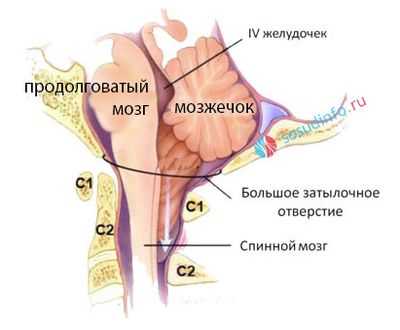
В продолговатом мозге сконцентрированы жизненно важные нервные центры, отвечающие за деятельность сердечно-сосудистой системы и дыхание, поэтому не только неврологический дефицит будет проявлением заболевания. В тяжелых случаях происходит угнетение витальных функций, и больной может умереть. Смещение гемисфер мозжечка влечет остановку циркуляции ликвора с гидроцефалией, которая еще больше усугубляет имеющиеся расстройства.
Аномалия Арнольда-Киари бывает врожденной, формирующейся у плода и сочетающейся с другими отклонениями в развитии, а клиника ее не всегда появляется сразу. В части случаев до манифестации патологии проходит значительный промежуток времени либо возникает ситуация, провоцирующая проявление бессимптомной до этого аномалии, у других пациентов и вовсе она может оказаться случайной находкой на МРТ. Нередко патология носит приобретенный характер и возникает под действием внешних причин, при этом мозг и череп при рождении имеют нормальное строение.
Причины и механизм развития порока задней черепной ямки (ЗЧЯ)
Единого мнения по поводу этиологии аномалии Киари нет. Ученые выдвигают различные теории, каждая из которых вполне обоснована и имеет право на существование.
Ранее аномалию считали исключительно врожденным пороком, однако наблюдения специалистов показали, что только малая часть пациентов имели дефекты во время внутриутробного развития, остальные же их приобрели уже в процессе жизни.
Причинами приобретенной патологии кранио-вертебрального перехода считают неравномерность скорости роста нервной ткани мозга и костной основы черепа, когда мозг увеличивается значительно быстрее, нежели костное вместилище, в котором он находится. Возникающее в конечном счете несоответствие объемов и служит основой болезни Арнольда-Киари.
Врожденная форма патологии сочетается с костной дисплазией, влекущей недоразвитие костей черепа, а также с нарушением формирования связочного аппарата, и любое внешнее воздействие, травма способны резко усугубить проявления патологии. Характерным считается сочетание порока черепной ямки с другими нарушениями утробного развития и врожденными синдромами.
Неврологами сформулировано два основных механизма формирования патологии:
- Уменьшение размеров ЗЧЯ при нормальных объемах отделов мозга (вероятно, вследствие нарушений во время внутриутробного периода).
- Увеличение объемов самого головного мозга при сохранении правильных параметров черепной ямки и большого затылочного отверстия, когда мозг оттесняет свои каудальные отделы в направлении затылочного отверстия.

Так как аномалия может быть врожденной, то среди причин указывают те, которые способны изменить нормальное течение беременности и внутриутробного развития:
- Злоупотребление медикаментами, прием алкоголя и курение при вынашивании плода, особенно — на ранних сроках, когда только формируются органы и системы зародыша;
- Вирусные поражения у беременных, среди которых особую опасность представляют инфекции с тератогенным эффектом — краснуха, цитомегаловирус и др.
Аномалия Арнольда-Киари возникает и по ряду приобретенных причин при изначально правильно развитых мозге и костях черепа. Привести к ее появлению уже после рождения способны:
- Родовые травмы, как спонтанные, так и при акушерских пособиях;
- Черепно-мозговые травмы и гидродинамический удар ликвора о стенки канала спинного мозга в случае нарушения ликвородинамики (так происходит у взрослых);
- Гидроцефалия.
Гидроцефалия может быть провоцирующим фактором, так как увеличение объема содержимого в черепе, пусть даже за счет жидкости, неминуемо влечет увеличение давления и смещение мозговых отделов в каудальном (заднем) направлении. С другой стороны, она является проявлением самой аномалии, когда опущение мозжечка вызывает блокаду ликворных путей и нарастание давления спинномозговой жидкости, циркулирующей по полостям мозга.
Типы и степени аномалии Арнольда-Киари
В зависимости от наличия тех или иных изменений со стороны мозга и костной основы черепа, принято выделять несколько разновидностей аномалии Арнольда-Киари:
- Аномалия Киари 1 типа, когда возникает перемещение книзу миндалин мозжечка, обычно выявляется у взрослых и подростков, нередко комбинируется с нарушением ликвородинамики и накоплением ликвора в центральном канале спинного мозга (гидромиелия). Возможно сдавливание мозгового ствола.
Аномалия Арнольда-Киари 1 типа - наиболее часто диагностируемая и имеющая довольно благоприятный прогноз
- Аномалия Арнольда-Киари 2 типа — манифестирует уже у новорожденных, так как происходит смещение значительно большего объема мозга, чем при 1 типе: миндалины мозжечка и его червь, продолговатый мозг с четвертым желудочком, возможно — образования среднего мозга. Обычно при 2 степени порока происходит нарушение тока ликвора с гидромиелией. Заболевание часто сочетается с наличием врожденной грыжи спинного мозга и аномалиями позвонков.
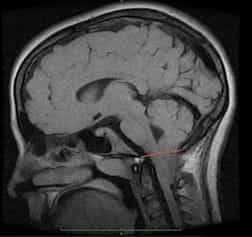
- 3 тип заболевания характеризуется выпячиванием мягкой мозговой оболочки с веществом мозга в затылочной области, куда попадают также мозжечок и продолговатый мозг.
аномалия Арнольда-Киари 3 типа на снимке
- Аномалия Арнольда-Киари 4 типа проявляется недоразвитием мозжечка, когда последний уменьшен, поэтому не опускается дистальнее к каналу в кости. Патология делает новорожденного нежизнеспособным и обычно заканчивается смертью.
Что касается степней тяжести, то:
Аномалию Арнольда-Киари 1 степени можно считать одним из наиболее легких вариантов патологии, так как пороков самого мозга при ней практически не возникает, а клиника может и вовсе отсутствовать, появляясь лишь при неблагоприятных условиях — травма, нейроинфекция и т. д.
Мальформации второй и третьей степени, в свою очередь, часто сочетаются с разнообразными пороками развития нервной ткани — гипоплазией некоторых участков мозга и подкорковых узлов, смещением серого вещества, кистами ликворных путей, недоразвитием извилин мозга.
Проявления синдрома Арнольда-Киари
Симптоматика синдрома Арнольда-Киари определяется его типом и характером смещения структур ЗЧЯ. Часто он протекает бессимптомно и обнаруживается случайно в ходе обследований мозга. У взрослых появление симптоматики может спровоцировать травма головы, у малышей некоторые формы заболевания заметны уже в первые часы и дни жизни.
Аномалия I типа диагностируется наиболее часто и может проявиться в подростковом либо взрослом возрасте следующими синдромами:
- Гипертензионным;
- Церебеллярным;
- Бульбарным;
- Сирингомиелическим;
- Явлениями поражения черепных нервов.
Гипертензионный синдром вызван увеличением внутричерепного давления вследствие блокады оттока ликвора сместившимися отделами мозга. Он проявляется:
- Головными болями в области затылка, особенно, при чихании, кашлевых толчках;
- Тошнотой и рвотой, после которой больной не ощущает облегчения;
- Напряжением мышц шеи.
Признаками вовлечения мозжечка (церебеллярный синдром) считают расстройства речи, двигательной функции, равновесия, нистагм. Пациенты жалуются на шаткость походки, неустойчивость положения тела в пространстве, затруднение мелкой моторики и четкости движений.
Поражение стволового отдела мозга представляется опасным ввиду расположения там ядер черепных нервов и жизненно важных нервных центров. Стволовая симптоматика состоит в:
- головокружении; двоении в глазах и снижении зрения;
- затруднении глотания;
- ухудшении слуха, шуме в ушах;
- обмороках, гипотонии, ночных апноэ.
Взрослые носители мальформации Арнольда-Киари указывают на нарастание головокружения и шума в ушах, а также пароксизмы потери сознания при поворачивании и наклонах головы. Вследствие сдавления стволов черепных нервов появляется атрофия половины языка и нарушение движения гортани с расстройством акта глотания, дыхания и голосообразования.
При образовании полостей и ликворных спинномозговых кист на фоне затрудненного тока спинномозговой жидкости у пациентов с I вариантом мальформации возникают признаки сирингомиелического синдрома — расстройство чувствительной сферы, онемение кожи, гипотрофия мышц, дисфункция тазовых органов, снижение и исчезновение брюшных рефлексов, периферические нейропатии и изменения со стороны суставов.
Расстройства чувствительности сопровождаются нарушением восприятия собственного тела, когда пациент, закрыв глаза, не может сказать, в каком положении находятся его руки или ноги. Снижается также чувствительность к боли и температуре.
По наблюдениям неврологов, диаметр и локализация кисты спинного мозга не обязательно отражаются на выраженности и распространенности нарушений чувствительной и двигательной сферы, гипотрофии мышц.
При синдроме второго и третьего типа течение патологии значительно тяжелее, симптоматика появляется у ребенка сразу же после родов. Характерны нарушения дыхания — стридор (шумное дыхание), приступы его остановки, а также двусторонний парез гортани, провоцирующий расстройства глотания, когда жидкая еда попадает в носовые ходы.
Второй тип аномалии у малышей первых месяцев жизни сопровождается нистагмом, усилением тонуса мышц в руках, синюшностью кожи, которые особенно заметны при кормлении грудничка. Двигательные нарушения вариабельны, проявления их меняются, возможна тетраплегия — паралич и верхних, и нижних конечностей.
Аномалия Арнольда-Киари третьего и четвертого вариантов протекает тяжело, это врожденная патология, которая не совместима с нормальной жизнедеятельностью, поэтому прогноз при таком диагнозе нельзя считать благоприятным.
Мальформация Арнольда-Киари может приводить к осложнениям, вызванным блокадой тока ликвора, поражением ядер черепных нервов, ущемлением стволовых структур. Самыми частыми считаются:
- Гипертензионно-гидроцефальный синдром — увеличение внутричерепного давления вследствие блокады путей ликворооттока, возможен как у детей, так и у взрослых;
- Расстройства дыхания, апноэ;
- Инфекционно-воспалительные процессы — бронхопневмонии, уроинфекции, которые связаны с лежачим положением пациента, нарушением актов глотания и дыхания, функции тазовых органов.
При тяжелом течении патологии может наступить кома, остановка сердца и дыхания, которые приводят к смерти в считанные минуты. Реанимационные мероприятия позволяют обеспечить витальные функции, но вернуть к жизни мозг и устранить необратимые последствия компрессии его отделов, к сожалению, практически невозможно.
Диагностика и лечение мальформации Арнольда-Киари
По особенностям симптоматики и на основании осмотра невролога диагноз мальформации Киари поставить невозможно. Энцефалография, исследования сосудов головы тоже не дадут никакой информации касательно причин неврологических нарушений, однако могут показать наличие повышенного давления в черепе. Рентгенография, КТ, МСКТ укажут на наличие дефектов костей черепа, которые характерны для этой патологии, но состояние мягкотканных структур, нервной ткани установить при этом нельзя.
Точная диагностика аномалии стала возможна благодаря использованию МРТ, посредством которой врач может определить и костные пороки, и варианты развития самого мозга, его сосудов, уровень расположения отделов относительно черепных костей, их размеры, объем задней черепной ямки и ширину большого затылочного отверстия. МРТ можно считать единственным точным и самым достоверным методом выявления патологии.
МРТ требует обездвиживания пациента, который должен какое-то время спокойно лежать на столе аппарата. У детей с этим могут возникнуть значительные трудности, поэтому исследование проводят в состоянии медикаментозного сна. Для поиска сочетанных пороков спинного мозга и позвоночника исследуют также эти отделы позвоночного столба.
Когда диагноз установлен, пациента направляют к нейрохирургу или неврологу для определения плана лечения, показаний к хирургической операции, ее вида.
Аномалия Арнольда-Киари, протекающая бессимптомно, не требует лечения. Более того, и сам носитель патологии может не догадываться о том, что в организме что-то не так. При появлении клинических признаков заболевания показано консервативное или оперативное лечение.
Если проявления ограничены головными болями, назначается медикаментозная терапия, включающая противовоспалительные средства (найз, ибупрофен, диклофенак), анальгетики (кеторол) и препараты, снимающие мышечный спазм (мидокалм).
При наличии неврологических расстройств, признаков сдавления отделов мозга, нервных стволов, в случае отсутствия эффекта от медикаментозного лечения в на протяжении 3 месяцев, больному необходима хирургическая коррекция.
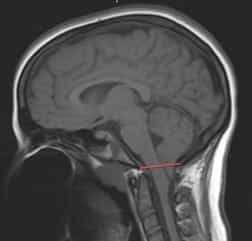
Операция необходима для устранения сдавливания нервной ткани и нормализации циркуляции ликвора. Самой популярной операцией при заболевании Киари считается краниовертебральная декомпрессия, которая направлена на увеличение размеров ЗЧЯ.
При декомпрессии хирург удаляет участки затылочной кости, резецирует миндалины мозжечка, по необходимости производит иссечение задних отделов первых шейных позвонков. Для предупреждения пролабирования задних частей мозга в полученное отверстие на твердую мозговую оболочку накладываются специальные синтетические заплаты.
пример удаления частей затылочкой кости и шейных позвонков
Декомпрессия ЗЧЯ считается и травматичной, и рискованной. Статистика такова, что осложнения возникают как минимум у каждого десятого больного, в то время как без операции показатель смертности намного ниже. В связи с высоким риском оперативного лечения нейрохирурги прибегают к нему только в случае действительно серьезных показаний — клиники сдавления участков мозга.
Другим вариантом хирургического лечения считается шунтирование, которое обеспечивает отток ликвора из полости черепа в грудную или брюшную полость. Путем имплантации специальных трубок происходит переток спинномозговой жидкости и снижение внутричерепного давления.
При тяжелых формах патологии показана госпитализация, мероприятия по профилактике инфекционных осложнений и коррекции неврологических расстройств. Нарастание отека мозга вследствие вклинивания его отделов в затылочное отверстие требуют лечения в условиях реанимации, включающего борьбу с отеком (магнезия, фуросемид, диакарб), налаживание искусственной вентиляции легких при нарушении дыхания и т. д.
Продолжительность жизни и прогноз при аномалии Арнольда-Киари зависят от типа патологии. При I типе прогноз можно считать благоприятным, в ряде случаев клиника не возникает совсем либо провоцируется лишь сильными травмирующими факторами. При бессимптомном течении носители аномалии живут столько же, сколько и все остальные люди.
При аномалии второго и первого типа с клиническими проявлениями прогноз несколько хуже, так как проявляется неврологический дефицит, который сложно устранить даже при активном лечении, поэтому для таких пациентов большое значение имеет своевременно проведенная операция. Чем раньше больному будет оказана хирургическая помощь, тем менее выраженные неврологические изменения его ждут.
Мальформация третьего и четвертого типов — наиболее тяжелые формы патологии. Прогноз неблагоприятен, так как вовлекаются многие структуры мозга, часто имеют место сочетанные пороки других органов, тяжелые нарушения функции ствола мозга, не совместимые с жизнью.
Читайте также:
- Алгоритм лечения при стабильной стенокардии. Комплексное лечечние стенокардии напряжения.
- Диагностика гемангиомы твердой мозговой оболочки и венозных синусов по КТ, МРТ
- Диагностика рс-вируса. Лечение поражений рс-вируса.
- Причины головокружения. Головокружение при патологии височной области
- Техника пальпации кишечника. Особенности пальпации кишок