Синдром Бартенверфера (Bartenwerfer) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синдром Жильбера: причины появления, симптомы, диагностика и способы лечения.
Определение
Синдром Жильбера (непрямая гипербилирубинемия), или семейная доброкачественная гипербилирубинемия носит наследственный характер и, соответственно, имеет пожизненное течение. Заболевание относится к хроническим патологиям печени и отличается чередованием периодов ремиссии и эпизодов желтухи, которые, как правило, провоцируются какими-либо факторами.
Синдром Жильбера манифестирует у подростков в период пубертата. У мужчин встречается в 3-4 раза чаще, чем у женщин.
Причины появления синдрома Жильбера
Непосредственной причиной развития синдрома Жильбера является нарушение обмена билирубина, вызванное мутацией в гене UGT1A1, который кодирует фермент (белок) уридиндифосфат (УДФ) - глюкуронилтрансферазу. Этот белок работает в клетках печени (гепатоцитах), а основная его функция - преобразование билирубина.
Выделяют две разновидности билирубина: прямой и непрямой.
И именно непрямой билирубин способен накапливаться в коже, вызывая ее желтую окраску.
Основным источником непрямого билирубина являются разрушенные красные кровяные тельца (эритроциты). У взрослого человека длительность жизни эритроцита составляет около 120 дней, после чего старые клетки разрушаются и им на смену приходят новые. Непрямой билирубин, высвобождающийся из разрушенного эритроцита, связывается с белком крови альбумином и переносится в печень. Здесь непрямой билирубин захватывается гепатоцитом, преобразуется при помощи УДФ- глюкоронилтрансферазы в прямой билирубин и выделяется из печени с желчью в просвет кишечника. Таким образом, вредный для организма непрямой билирубин обезвреживается и выводится.

У людей, страдающих синдромом Жильбера, нарушается процесс преобразования билирубина, в результате чего у них умеренно повышается уровень непрямого билирубина. В большинстве случаев заболевание не представляет опасности для здоровья человека, но вызывает пожелтение кожи, слизистых и склер глаз.
Классификация заболевания
Как таковой классификации синдрома Жильбера не существует. Однако для врача становится критически важным исключить другие виды желтухи, которые могут быть признаками значительно более грозных заболеваний, нежели синдром Жильбера.
По причине развития выделяют:
- желтухи гемолитические,
- желтухи печеночные,
- желтухи механические.
Синдром Жильбера относится к печеночному типу желтухи, поскольку связан с нарушением работы печени по утилизации непрямого билирубина.
Симптомы синдрома Жильбера
Для заболевания характерно пожелтение кожи, слизистых и склер глаз, которое может иметь различную степень выраженности и носит волнообразный характер.
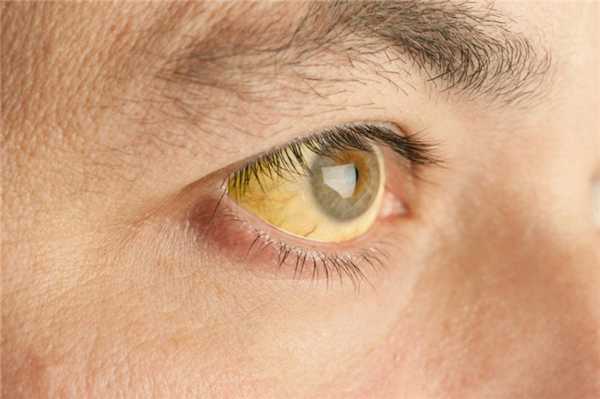
Эпизоды обострения могут быть спровоцированы различными внешними и внутренними факторами. Типичные эпизоды желтухи могут сопровождаться тяжестью и болями в правом подреберье, что связано с нарушением оттока желчи из-за изменения ее состава. По той же причине отмечаются признаки нарушения пищеварения, например, изменение характера стула, появление отрыжки, усиление газообразования. В некоторых случаях наблюдаются астеновегетативные проявления - подавленное настроение, быстрая утомляемость, плохой сон, головокружение.
Диагностика синдрома Жильбера
При развитии у пациента желтухи наряду с клиническим анализом крови с подсчетом лейкоцитарной формулы назначают биохимический анализ крови.
Синонимы: Общий анализ крови (ОАК); Гемограмма; КАК; Развернутый анализ крови. Full blood count; FBC; Complete Blood Count (CBC); Hemogram; CBC with White Blood Cell Differential Count; Peripheral Blood Smear; Blood Film Examination; Comp.
Синдром Барттера
Синдром Барттера - это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.

Общие сведения
Синдром Барттера в клинической урологии представляет собой редкую генную мутацию - дефект петли Генле, наследуемую по аутосомно-рецессивному типу и проявляющуюся, как правило, уже в детском возрасте. Неспособность почечных нефронов задерживать калий приводит к хронической потере его с мочой и уменьшению объема циркулирующей крови при нормальном или пониженном АД. В зависимости от вида пораженных генов различают: неонатальный синдром Барттера 1 и 2 типов, классический синдром Барттера, синдром Гительмана. Также встречается приобретенный синдром псевдо-Барттера, характеризующийся сходными проявлениями, но не сопровождающийся патологией почечных канальцев.
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагностика
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике - сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Синдром Барттера — это заболевание почек
Синдром Барттера (тубулопатия) — это заболевание почек, которое относится к врожденным дефектам. Заболевание наследуется от обоих родителей. Это нарушение кислотно-щелочного баланса в организме в результате плохой работы почечных спиралей. Нарушения в фильтрации крови приводят к недостатку калия и натрия в организме.
Синдром Барттера — это группа генетически обусловленных заболеваний, называемых тубулопатиями. Это заболевания, связанные с нарушением резорбтивной или секреторной функции почечных канальцев, возникающие при нормальной или только минимально сниженной клубочковой фильтрации. Синдром Барттера чаще всего встречается в возрасте от одного до двух лет. Это редкое заболевание, и оно наследуется рецессивно, то есть ребенок должен получить по одной копии дефектного гена от каждого родителя. Мутации связаны с четырьмя генами, связанными с кодированием белка, ответственными за реабсорбцию.
При синдроме Барттера встречаются следующие симптомы:
- гипокалиемический нарушение кислотно-щелочного баланса (алкалоз)
- высокий уровень гормонов в плазме, называемых ренином и альдостероном
- низкое кровяное давление
- артериальная нечувствительность к ангиотензину, то есть гормон, который регулирует ионы натрия и калия.
Характеристика заболевания
Заболевание представляет собой нарушение реабсорбции натрия, калия и хлорида в клубочек, в Петля Генле, которая влечет за собой изменения в составе крови и проблемы с выработкой мочи. Клубочки — это сеть правильно построенных капилляров, через которые фильтруется кровь, вода и питательные вещества, что приводит к первичной мочи. Любое нарушение работы почек на этой стадии приводит к слишком большой потере ионов калии, натрии и хлора. Когда они выводятся из организма в чрезмерных количествах, существующий кислотно-щелочной баланс перестает существовать. Речь идет о метаболический алкалоз, то есть состояние повышенного pH в плазме крови, вызванное потерей ионов калия.
Что происходит потом? Когда клубочки истощают слишком много ионов калия и натрия, и в организме начинает не хватать циркулирующей крови, организм реагирует, увеличивая выработку гормонов — ренина, ангиотензина и альдостерона. Обычно этих соединений мало в организме, и они ответственны за снижение потери воды и кровяного давления. Но сейчас их избыток приводит к развитию болезни.
Синдром Барттера встречается в нескольких вариантах, в зависимости от того, какая комбинация генов повреждена и, следовательно, какой ионный канал не работает должным образом. Отдельные виды заболеваний, в том числе синдром Гительмана немного отличается по ходу, но общим для них является снижение уровня калия в крови (так называемая гипокалиемия). При одном из вариантов заболевания слух также может быть поврежден.
Симптомы
- полиурия, то есть частое мочеиспускание с возможностью обезвоживания
- чрезмерная жажда
- низкое кровяное давление
- замедление роста и наращивания веса
- тенденция к развитию камней в почках
- запор
- усталость, мышечная слабость
- осложнения, такие как сердечные заболевания, остеопороз.
Диагностика и лечение
Врожденный синдром Барттера диагностируется у очень маленьких детей с помощью диагностических тестов. Вы также можете видеть, что ребенок набирает вес слишком медленно и плохо растет. Генетическое тестирование также может быть сделано, чтобы точно определить, какие гены были повреждены.
Как генетический дефект, синдром Барттера неизлечим. Однако ведется консервативное лечение. Он заключается в пероральном введении препаратов калия и пополнении уровня других электролитов в организме, в некоторых случаях даже внутривенно. В менее тяжелых состояниях это достаточно, чтобы больной вел достаточно нормальную жизнь. Более тяжелые случаи заболевания, когда возникает почечная недостаточность, требуют диализа и даже трансплантации почки.
Синдром Жильбера - симптомы и лечение
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Жильбера — это генетический пигментный гепатоз с аутосомно-доминантным типом наследования, протекающий с повышением уровня неконъюгированного (свободного) билирубина, чаще проявляющееся в период полового созревания и характеризующийся доброкачественным течением [1] .
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
К провоцирующим факторам проявления синдрома можно отнести:
- голодание или переедание;
- жирную пищу;
- некоторые лекарственные средства;
- алкоголь;
- инфекции (грипп, ОРЗ, вирусный гепатит);
- физические и психические перегрузки;
- травмы и оперативные вмешательства.
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
- желтуха различной степени выраженности;
- ксантелазмы век (жёлтые папулы);
- периодичность появления симптомов [1] .
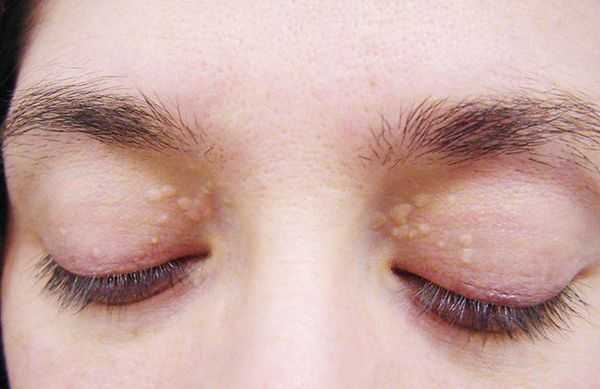
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
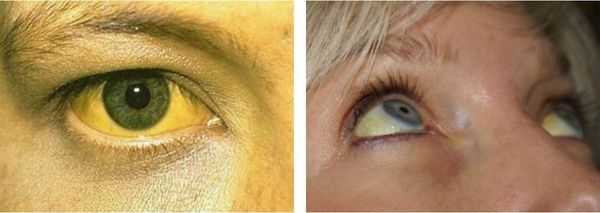
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
- слабость;
- недомогание;
- подавленность;
- плохой сон;
- снижение концентрации внимания.
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
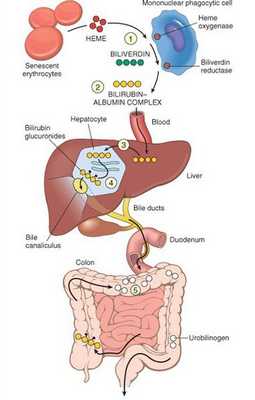
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами ("жирами") клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
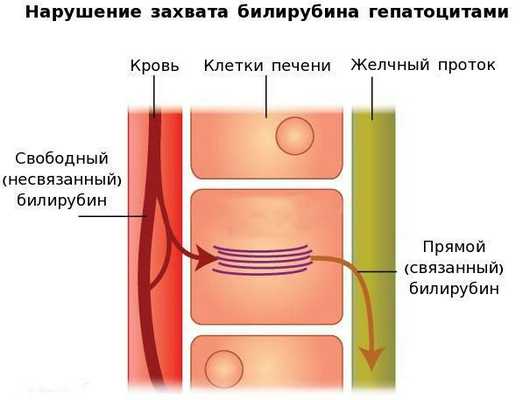
В конечном итоге вышеперечисленные патологические процессы приводят к увеличению содержания свободного (несвязанного) билирубина в плазме, что обуславливает клинические проявления заболевания [6] .
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Синдром Жильбера: причины, симптомы, диагностика, лечение
Синдром Жильбера - самая распространенная форма наследственного пигментного гепатоза, характеризующаяся нарушением обмена билирубина вследствие генетического дефекта микросомальных ферментов печени. Часто заболевание протекает бессимптомно многие годы, но под действием провоцирующих факторов могут возникать интермиттирующая желтуха, диспепсические и астеновегетативные расстройства.
Диагностика требует сбора семейного анамнеза, проведения лабораторных и инструментальных исследований. В лечении синдрома Жильбера применяются медикаментозные препараты разных групп.
Синдрому Жильбера по МКБ соответствует код E80.4. Мужчины страдают данным заболеванием в четыре раза чаще женщин. В целом же распространенность патологии в европейских и азиатских странах не превышает 5%.
Причины появления
В норме у человека при распаде эритроцитов образуется токсичное для организма вещество - непрямой билирубин. Он нейтрализуется клетками печени, которые обеспечивают его связывание с глюкуроновой кислотой и превращение в водорастворимый прямой билирубин. В дальнейшем происходит выведение данного соединения через органы билиарной системы с калом и мочой.
Причина возникновения врожденного синдрома Жильбера - генетическая мутация микросомального фермента УДФ-глюкуронилтрансферазы, ответственного за связывание глюкуроновой кислоты с непрямым билирубином. Вторым механизмом развития болезни считается нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой. Таким образом, концентрация токсичных продуктов метаболизма в сыворотке крови растет. В результате неконъюгированный билирубин накапливается в тканях, окрашивая их в желтый цвет. Наследование патологии происходит по аутосомно-доминантному типу.
- несбалансированное питание, строгая диета или голодание;
- потеря жидкости организмом, обезвоживание;
- стресс, физическое переутомление;
- менструальное кровотечение;
- инфекционные процессы;
- операции;
- прием алкоголя и некоторых лекарственных препаратов.
Первые проявления синдрома Жильбера у детей начинаются в подростковом возрасте, после 11-12 лет, что связано с угнетающим влиянием половых гормонов на утилизацию билирубина. Однако дебют заболевания может случиться и намного позже, в срок до 30 лет.
- Интермиттирующая желтуха. Локализация и степень выраженности могут быть различными - от изменения цвета только склер глазных яблок до яркой желтушности кожи и слизистых по всему телу. Появление желтухи носит всегда внезапный характер, возникает после воздействия провоцирующих факторов и самопроизвольно исчезает.
- Ксантелазмы век. Единичные или множественные бляшки желтоватого цвета, возвышающиеся над кожей.
- Ощущение тяжести в правом подреберье, чувство дискомфорта в брюшной полости. Данные симптомы связаны с увеличением печени, селезенки, развитием холецистита, дискинезии желчевыводящих путей, желчнокаменной болезни.
- Астеновегетативные расстройства. Из-за интоксикации организма появляются быстрая утомляемость и подавленность, нарушение сна, избыточное потоотделение.
- Диспепсические проявления. Потеря аппетита, привкус горечи во рту, тошнота, отрыжка, вздутие живота и избыточное газообразование, нарушения стула.
Осложнения
Даже при благоприятном течении заболевания пациенту важно знать, чем опасен синдром Жильбера. При его наличии возрастает риск развития желчнокаменной болезни, внутрипеченочного и внепеченочного холестаза, дискинезии желчевыводящих путей, а также существенно отягощается течение других патологий гепато-билиарной системы, например, лекарственных и токсических гепатитов. Кроме того, развитие желтухи сопровождается снижением иммунитета.
Диагностика синдрома Жильбера
Постановка диагноза включает анализ имеющихся жалоб и клинических проявлений заболевания, уточнение наличия эпизодов желтухи и патологии гепато-билиарной системы у близких родственников, применение лабораторных и инструментальных методов исследования.
Дифференциальная диагностика синдрома Жильбера проводится с вирусным и хроническим гепатитом, механической и гемолитической желтухой, синдромом Криглера - Найяра, синдромом Дабина - Джонсона и синдромом Ротора, первичной шунтовой гипербилирубинемией, врожденными циррозами печени, атрезией желчных ходов. Главная отличительная особенность данного нарушения - повышение фракции неконъюгированного билирубина в сыворотке крови, не связанное с гемолизом эритроцитов, и наследственная отягощенность.
- . В ряде случаев отмечается повышение уровня гемоглобина или снижение числа тромбоцитов. . Основной признак заболевания - повышение концентрации общего билирубина до 34,2-85,5 мкмоль/л за счет его непрямой фракции. Показатели остальных печеночных ферментов крови, таких как АсАТ, АлАТ, ГГТП, ЩФ, как правило, находятся в пределах нормы. Возможно наличие диспротеинемии и повышение концентрации общего белка. . Проводится определение креатинина. . При синдроме Жильбера наличие Ig G к HBcAg, гепатиту А и С нехарактерно. . Анализ кала на стеркобилин при рассматриваемой патологии имеет отрицательный результат. В сложных случаях показано проведение функциональных лабораторных проб. Снижение уровня билирубина на фоне приема фенобарбитала и его повышение в 1,5-2 раза после голодания, низкокалорийной диеты или внутривенного введения никотиновой кислоты подтверждают диагноз.
Из инструментальных методов наиболее часто выполняется ультразвуковое исследование органов брюшной полости для исключения других патологий печени и желчнокаменной болезни. У детей можно пользоваться способом определения печеночно-селезеночного индекса по данным УЗИ.
Дополнительно возможно проведение дуоденального зондирования, тонкослойной хроматографии. Чрезкожная пункция печени с последующей морфологической оценкой полученного биоматериала выполняется при подозрении у пациента хронического гепатита или цирроза печени.
Лечение синдрома Жильбера
Само по себе наличие у детей и взрослых синдрома Жильбера еще не является показанием к медикаментозному лечению. Для предупреждения обострения заболевания и развития осложнений важны общие мероприятия: ограничение занятий тяжелыми видами спорта, продолжительного пребывания на солнце, употребления алкогольных напитков и гепатотоксических препаратов. Таким пациентам необходимо правильно питаться, избегать длительного голодания, а также продуктов, содержащих трудноусвояемые жиры - жирных сортов мяса, жареных и острых блюд, консервированной пищи.
- прием индукторов микросомальных ферментов (фенобарбитал, зиксорин);
- прием активированного угля;
- фототерапию.
Прогноз заболевания в любом возрасте благоприятный. Гипербилирубинемия при синдроме Жильбера сохраняется пожизненно, но носит доброкачественный характер, не сопровождается прогрессирующими изменениями в печени и не оказывает влияние на продолжительность жизни.
Читайте также:
- Специализированная медицинская помощь при ожоге. Дезинтоксикационная терапия при ожоговой токсемии.
- Симптомы агранулоцитоза в полости рта и его лечение
- Повышенная стираемость зубов: причины, симптомы и лечение
- Общие сведения об инфекционных заболеваниях
- Боевые отравляющие вещества удушающего действия