Синдром Берчи-Роше (Bartschi-Rochaix) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 02.02.2026
Синдром Рокитанского-Кюстнера-Майера-Хаузера - патология, которая развивается у женщин с нормальным кариотипом (46XX) вследствие аномалии развития протоков Мюллера и представляет собой полное отсутствие или недоразвития матки, маточных труб и влагалища.
Причины и симптомы
На 10-12 неделе развития эмбриона женского пола мюллеровы протоки начинают преобразовываться во внутренние половые органы:
- Верхний отдел протоков Мюллера образует фаллопиевы трубы
- Средняя часть сливается и формирует тело и шейку матки
- Нижний отдел образует влагалище (верхнюю его часть)
Однако очень редко (примерно в 0,5% случаев) этот процесс нарушается, приводя к аномалиям развития тела и шейки матки, а также 2/3 верхней части влагалища.
О том, что у женщины нет матки, она, как правило, узнает в подростковом возрасте, обратившись к гинекологу с жалобами на отсутствие менструаций (первичную аменорею). При этом пациентки с синдромом Рокитанского-Кюстнера имеют нормальный кариотип (46XX) и наружные половые органы. Яичники имеют неизмененную структуру и функцию, в связи чем у пациенток с аплазией матки имеются хорошо развитые вторичные половые признаки. Гормональный фон соответствует двухфазному овуляторному менструальному циклу, а некоторые женщины испытывают характерные циклические изменения (нагрубание молочных желез, боли в нижней части живота).
Синдром Рокитанского-Кюстнера, как правило, имеет спорадический (случайный) характер, однако фиксировались и несколько случаев в пределах одной семьи.
Анализируя семейный анамнез, специалисты пришли к выводу, что при наследовании синдрома имеет место аутосомно-доминантный тип. До сих пор ученым не удалось выяснить, мутация в каких именно генах приводит к синдрому Рокитанского-Кюстнера, хотя существуют гипотезы относительно некоторых генов, расположенных в различных хромосомах.
Спорадические случаи, по мнению специалистов, могут быть обусловлены рядом факторов:
- Дефект развития мезодермальной паренхимы (зародышевой соединительной ткани)
- Воздействие эндогенных и экзогенных тератогенных факторов
- Недостаток выработки биологически активной субстанции MIS, необходимой для нормального развития мюллеровых протоков
- Полное отсутствие или недостаточное количество рецепторов эстрогенов в мюллеровых протоках (их нижнем отделе) на определенных этапах эмбриогенеза
Диагностика
- Сбор анамнеза, оценка развития вторичных половых признаков
- Бимануальный смотр гинеколога, в ходе которого выявляется отсутствие матки. Влагалище представляет собой короткий слепой отросток длиной до 15 мм.
- УЗИ органов малого таза, демонстрирующее отсутствие (нарушение развития) матки и фаллопиевых труб, а также наличие яичников
- Гормональное исследование, подтверждающее нормальную функцию яичников
В ходе диагностики синдром Рокитанского-Кюстнера должен быть дифференцирован с синдромом текстикулярной феминизации (одной из форм мужского псевдогермафродитизма, когда мужской генотип XY сочетается с женским фенотипом), изолированной атрезией (сращением стенок) влагалища.
Лечение
Лечение синдрома Рокитанского-Кюстнера направлено на восстановление половой функции и купирование болевого синдрома, обусловленного скоплением менструальной крови в брюшной полости (при недоразвитии матки).
Для нормализации половой жизни пациентки в большинстве случаев рекомендован кольпопоэз (создание искусственного влагалища) с использованием фрагмента сигмовидной кишки.
Преодоление бесплодия, обусловленного аплазией матки
Женщины, страдающие синдромом Рокитанского-Кюстнера, не в состоянии самостоятельно выносить беременность. Однако они имеют нормально функционирующие яичники, в которых созревают яйцеклетки. В связи с этим для рождения генетически родного ребенка применяется метод суррогатного материнства: эмбрионы, полученные после оплодотворения яйцеклеток пациентки спермой ее мужа, переносят в полость матки суррогатной матери. Пункция яичников осуществляется в этом случае либо лапароскопическим путем, через прокол брюшной стенки, либо через свод неовлагалища, если предварительно был проведен кольпопоэз.
Специалисты Нова Клиник имеют большой успешный опыт проведения программ суррогатного материнства у пациенток с синдромом Рокитанского-Кюстнера-Майера, включающий полное медицинское и юридическое сопровождение процедуры вплоть до получения свидетельства о рождении ребенка в органах ЗАГС.
БАРШОНЯ-ТЕШЕНДОРФА СИНДРОМ
БАРШОНЯ-ТЕШЕНДОРФА СИНДРОМ (Th. Barsony, W. Teschendorf; синоним: множественные ложные дивертикулы, множественные функциональные дивертикулы, четкообразный пищевод, извитой пищевод, штопорообразный пищевод) — заболевание пищевода, характеризующееся множественными циркулярными спастическими сокращениями его стенок.
Этиология Баршоня — Тешендорфа синдрома неизвестна. Заболевание встречается преимущественно в пожилом возрасте. Клинически отмечаются дисфагия, часто непостоянная, иногда боли за грудиной, симулирующие стенокардию.
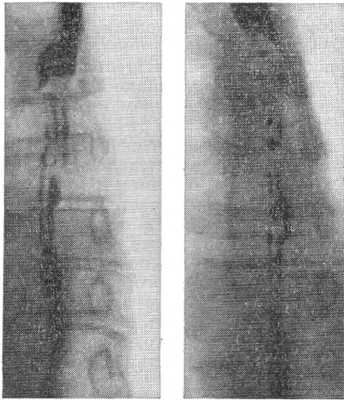
Синдром Баршоня — Тешендорфа: изменчивость рентгенологической картины пищевода (снимки сделаны с интервалом в 15 мин.).
Диагностика возможна лишь с помощью контрастного рентгенологического исследования, при котором определяется характерный четкообразный вид пищевода. Участки пищевода, расположенные между двумя соседними спастически сокращенными отрезками, могут иметь округлую, овоидную или неправильную косую форму. Нередко можно наблюдать спонтанное расправление спазмированных участков пищевода или перемещение их в каудальном направлении; в других случаях рентгеновская картина длительное время остается стабильной.
Применение атропина, папаверина, метацина, нитритов приводит к временному ослаблению или исчезновению спазмов и восстановлению проходимости пищевода. Изменчивость рентгенологической картины (рис.), а также реакция на указанные препараты позволяют правильно провести дифференциальную диагностику. В трудных для диагностики случаях может быть применена париетография пищевода (см. Париетография).
Прогноз благоприятный.
Библиогр. Каган E. М. Рентгенодиагностика заболеваний пищевода, М., 1968; Розенштраух JI. С. и Демин В. А. Комплексное рентгенологическое исследование при раке пищевода, в кн.: Вопр, рентгенодиагностики, под ред. Л. С. Розенштрауха, кн. 1, с. 145, М., 1964; T а г e р И. Л. Рентгенологическое исследование при дисфагии, М., 1947, библиогр.; Фанарджян В. А. Рентгенодиагностика заболеваний пищеварительного тракта, т. 1, Ереван, 1961; Birsony Т. Funktionelle Speiserohren-Divertikel (Relaxations-Divertikel), Wien, klin. Wschr., S. 1363, 1926; Teschendorf W. Die Rontgenuntersuchung der Speiserohre, Ergebn. med. Strahlenforsch., Bd 3, S. 175, 1928, Bibliogr.
Синдром Туретта - симптомы и лечение
Что такое синдром Туретта? Причины возникновения, диагностику и методы лечения разберем в статье доктора Диордиева Максима Борисовича, психиатра со стажем в 8 лет.
Над статьей доктора Диордиева Максима Борисовича работали литературный редактор Вера Васина , научный редактор Владимир Вожжов и шеф-редактор Маргарита Тихонова

Определение болезни. Причины заболевания
Синдром Туретта (Tourette's syndrome) — это заболевание нервной системы, при котором возникают множественные двигательные и вокальные тики. Для постановки диагноза они должны присутствовать дольше года.

Впервые заболевание, похожее на синдром Туретта, было описано в 1486 году в книге «Молот ведьм». Там упоминался священник с моторными и вокальными тиками, считавшийся одержимым. В конце XIX века симптомы заболевания на примере нескольких пациентов описал вместе с коллегами французский невролог Жорж Жиль де ла Туретт, в честь которого и назван синдром [1] .
Обычно синдром Туретта проявляется уже в детстве, но часто заболевание выявляют поздно или не диагностируют вовсе, так как родители не обращают на тики должного внимания. Из-за этого маленькие пациенты не получают своевременной помощи и могут страдать не только от самих тиков, но и от психологических проблем, связанных с заболеванием. В Европе от возникновения первых симптомов синдрома Туретта до постановки диагноза проходит в среднем более 5 лет [2] .
Распространённость синдрома Туретта
Синдром Туретта очень распространён — он встречается примерно у 10 из 1000 детей. В России его диагностируют у 8 человек на 10 000 населения, им могут страдать до 5 % школьников [3] . Мужчины болеют чаще, чем женщины: соотношение между ними составляет примерно 3 к 1.
Причины синдрома Туретта
Синдром Туретта — это генетическое расстройство, которое передаётся от родителей. Однако точный механизм наследования и ген, ответственный за болезнь, не известны. Риск передачи заболевания ребёнку составляет около 50 %. В прошлом, в начале XX века, тики считались следствием психотравм, но современная медицина это отвергает, так как такое предположение не удалось доказать [4] . Психосоциальные факторы и аутоиммунные заболевания не являются причиной синдрома Туретта, но могут влиять на тяжесть течения болезни.
Существует теория, что недостаток магния в организме и связанные с ним нарушения обмена веществ могут влиять на развитие синдрома Туретта. Косвенным доказательством этого служит то, что препараты с некоторыми соединениями магния могут улучшать состояние больных. Однако большие исследования на эту тему не проводились [5] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Туретта
Синдром Туретта проявляется тиками — быстрыми, внезапными, повторяющимися навязчивыми движениями или произнесением звуков. Чаще всего они возникают у мальчиков в возрасте от 4 до 11 лет. Тяжесть тиков доходит до пика примерно в 10-12 лет и ослабляется в подростковом возрасте. Большинство тиков исчезают спонтанно, но примерно у 1 % детей они сохраняются во взрослой жизни [7] .
Выделяют две основные группы тиков:
- Моторные тики — это непроизвольные движения частей тела. Самый распространённый из них — усиленное моргание. Также могут возникать подпрыгивания, постукивания по себе, развороты и повороты тела, гримасы, плевки, нецензурная жестикуляция и повторения чужих движений (копропраксия и эхопраксия).
- Вокальные, или звуковые, тики — это навязчивое произношение звуков, реже слов. Может проявляться кашлем, покашливанием, кряхтением. Иногда таких детей ошибочно лечат от бронхитов, трахеитов и бронхиальной астмы. Синдром Туретта часто ассоциируется с копролалией — внезапным высказыванием нецензурных фраз или слов, которое зачастую сопровождается копропраксией. Однако копролалия возникает только у 10 % пациентов [6] . Помимо копролалии, они могут повторять чужие слова, собственное слово или фразу (эхолалия и палилалия).
При синдроме Туретта моторные тики обязательно сочетаются с вокальными. Если присутствуют моторные тики, но нет вокальных, то стоит заподозрить другие заболевания: органическое поражение головного мозга, эпилепсию, синдром дефицита внимания (СДВГ), обсессивно-компульсивное расстройство (ОКР).
У многих детей и подростков с синдромом Туретта также отмечается СДВГ, ОКР, повышенная агрессивность, тревожность и склонность к депрессиям.
При синдроме Туретта интеллектуальные способности не нарушаются. Дети с этим заболеванием могут сильно расстраиваться от подшучиваний своих сверстников. При эмоциональном напряжении, вызванном пристальным вниманием или насмешками окружающих, тики могут усиливаться.
Форма тиков при синдроме Туретта может меняться в течение суток или недели, например от лёгких единичных моторных тиков утром или в начале недели до сложных и множественных по вечерам или под конец учебной недели. Видимо, их выраженность зависит в том числе от психоэмоциональных нагрузок.
Иногда дети пытаются сдерживать тики, но такой контроль возможен лишь в некоторой степени. Когда ребёнок старается подавить тики, симптомы могут усилиться. Попытка сдержать тик вызывает выраженный дискомфорт, из-за чего возрастает тревога — тикозные движения, наоборот, немного успокаивают. Из-за стресса, тревожных состояний и усталости тики могут учащаться и усиливаться.
Патогенез синдрома Туретта
Патогенез синдрома Туретта до конца не изучен. Известно лишь, что расстройство вызвано генетическими причинами. Скорее всего, при определённых генетических факторах нарушается работа нейромедиаторных систем в подкорковых образованиях и лобной коре.
Помимо генетических факторов, в патогенезе может участвовать и органическое повреждение головного мозга, например при патологии беременности и родов, черепно-мозговых травмах или нейроинфекциях.
Основная роль в патогенезе заболевания, вероятно, принадлежит дисфункции лобных долей. Считается, что большую роль в развитии синдрома Туретта играет правая лобно-височная область, сенсомоторные отделы орбитофронтальной коры, моторная область, базальные ганглии и поясная извилина. Также важное значение имеют нарушения в кортико-стрио-таламо-кортикальном контуре — нейронных цепях, связывающих кору, базальные ганглии и таламус.

Нарушения в работе этих структур также характерны для детей с ОКР и СДВГ — эти заболевания часто сопутствуют синдрому Туретта. Даже известны генетически связанные с синдромом Туретта формы ОКР (преимущественно ОКР с навязчивыми действиями — F42.1).
Предположительно, при синдроме Туретта нарушается работа дофаминергической системы. Изменения, вероятно, затрагивают серотонин-, норадреналин-, глутамат-, холин-, ГАМКергическую и опиоидную системы. Косвенно на связь синдрома Туретта с дофамином указывает то, что тики уменьшаются при лечении препаратами, которые воздействуют на передачу нервного импульса, вызванную дофамином ( например, путём блокады постсинаптических D2-рецепторов) [2] .
Классификация и стадии развития синдрома Туретта
Синдром Туретта — это разновидность хронических тиковых (или тикозных) нарушений. В Международной классификации болезней (МКБ-10) заболевание кодируется как F95.2 Комбинированные голосовые и множественные двигательные тики.
В следующей Международной классификации болезней (МКБ-11) тики и синдром Туретта из психических расстройств перенесены в неврологические [1] .
Согласно классификации Американской психиатрической ассоциации (DSM-IV), тики подразделяются на следующие группы:
- по виду — двигательные или голосовые;
- по продолжительности — преходящие или хронические.
Преходящее тиковое расстройство — это множественные двигательные, голосовые или тики обоих видов, которые длятся от 1 до 12 месяцев. Хронические тиковые расстройства присутствуют больше года. Они могут быть одиночными или множественными, двигательными или голосовыми, но не оба вида сразу. Синдром Туретта относится к хроническому тиковому расстройству. Для постановки диагноза необходимо, чтобы множественные двигательные тики и хотя бы один голосовой тик наблюдались более года.
Стадии синдрома Туретта не выделяют. Но обычно расстройство начинается с преходящих двигательных тиков, как правило подёргивания лица, которые длятся до года. Часто это гримасничание, затем покашливание и шипение. Постепенно тики распространяются на руки, ноги и мышцы шеи. Затем, обычно через год, присоединяются вокальные тики, которые осложняют картину болезни.
Осложнения синдрома Туретта
Синдром Туретта может сопровождаться депрессией, тревожным расстройством, ОКР и СДВГ, что осложняет прогноз. Депрессия возникает из-за того, что детей с этим синдромом часто обижают и унижают. В результате у них формируется чувство одиночества и может развиться аутоагрессия, вплоть до попытки суицида. Поэтому важно не оставлять ребёнка один на один с этим расстройством: ему особенно необходима поддержка родителей и друзей.
При симптоме копролалии дети выкрикивают нецензурную брань, из-за чего могут подвергаться агрессии со стороны окружающих. Поэтому для таких пациентов очень важно организовать правильную социальную среду [7] .
Диагностика синдрома Туретта
Синдром Туретта диагностирует врач-психиатр или невролог, основываясь на наблюдении и сборе сведений об истории болезни, условиях жизни и перенесённых заболеваниях. Сейчас разрабатываются генетические карты, которые позволят с самого рождения определять совокупность генов, характерных для синдрома Туретта, но пока этот метод недоступен.
Чтобы установить диагноз «синдром Туретта», состояние должно соответствовать следующим критериям:
- присутствуют множественные двигательные тики и как минимум один голосовой тик;
- тики возникают много раз в день, почти ежедневно;
- расстройство длится более года, но необязательно непрерывно, ремиссии продолжаются меньше двух месяцев;
- симптомы появились в возрасте до 18 лет [8] .
Очень важно отличать тики при синдроме Туретта от вторичных тиков, которые появились на фоне инфекций или черепно-мозговых травм при беременности, родах или в раннем детстве. Их различают только на основе анамнеза и физикального обследования. Другие методы, например магнитно-резонансная томография и анализы крови, для диагностики синдрома Туретта не используются.
Лечение синдрома Туретта
Перед врачом всегда стоит выбор — назначать ли препараты при синдроме Туретта. При этом важно ориентироваться на состояние пациента, так как более чем в половине случаев симптомы исчезают без приёма медикаментов.
Психологическая помощь
Если тики не мешают человеку общаться, учиться и работать, то, скорее всего, принимать препараты не нужно. В такой ситуации будет полезно психологическое консультирование ребёнка и родителей. Важно рассказать родителям, что от ребёнка ни в коем случае нельзя требовать, чтобы он перестал кашлять и гримасничать, — это только усилит эмоциональное напряжение и, соответственно, тики.
В зарубежных странах, особенно в США, хорошо зарекомендовал себя метод под названием «Habit reversal training», т. е. тренировка отмены привычки [10] . Пациента учат отслеживать ощущение, предшествующее тикам, и пытаться заменить их на более приемлемые действия. Метод не избавляет от тиков, но заметно уменьшает их проявления.
Также для коррекции поведения используется когнитивно-поведенческая психотерапия [12] .
Медикаментозное лечение
Для лечения синдрома Туретта могут применяться антипсихотики (нейролептики) в невысоких дозировках, которые воздействуют на дофаминэргическую активность. Наиболее эффективны Арипипразол, Рисперидон и Галоперидол. Из них лучше всего переносится Арипипразол. Однако в российских инструкциях по лечению синдрома Туретта это лекарство не упоминается, хотя в США оно одобрено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) и активно используется в терапии. В России назначают Галоперидол и Рисперидон, а также ряд ноотропов (чаще всего гопантеновую и аминофенилмасляную кислоту) без достаточной доказательной базы.
Есть данные об эффективности Клонидина, но этот препарат опасен при передозировке. Среди возможных побочных эффектов — коллаптоидные состояния, т. е. резкое падение артериального давления, которое может привести к развитию обморока.
Существуют не подтверждённые данные об эффективности инъекции ботулотоксина в мышцы лица.
При возникновении сопутствующих заболеваний, таких как тревожно-депрессивное расстройство и ОКР, применяются селективные ингибиторы обратного захвата серотонина: Сертралин, Пароксетин, Эсциталопрам. При выраженной агрессии, направленной на себя или окружающих, назначаются нормотимики (препараты лития и другие) [12] .
Транскраниальная магнитная стимуляция
В настоящее время изучается влияние транскраниальной магнитной стимуляции на синдром Туретта как у детей, так и у взрослых, но пока недостаточно данных об эффективности этого метода [11] .
Нейрохирургическое лечение
При тяжёлом течении заболевания и выраженной устойчивости к медикаментам может применяться нейрохирургический подход — глубокая стимуляция подкорковых структур головного мозга (бледного шара и таламуса). Её используют для взрослых пациентов.
Прогноз. Профилактика
Прогноз благоприятнее и заболевание чаще заканчивается ремиссией или выздоровлением, если тики появились в возрасте до 7 лет.
Синдром Туретта часто вызывает у людей страх и ассоциации, что эта особенность мешает нормально общаться с другими. Но если грамотно подойти к терапии и исключить провоцирующие факторы, расстройство может протекать вполне благоприятно. Примером служит певица Билли Айлиш. Из-за тяжести заболевания она не ходила в школу и обучалась дома. Это не помешало певице в 2019 году записать сингл, завоевавший первые места в мировых хит-парадах, а в 2021 году войти в список 100 наиболее влиятельных людей года по версии журнала Time. Также синдромом Туретта, предположительно, страдал Вольфганг Моцарт [13] .
Синдром Туретта — это генетическое заболевание, поэтому предупредить его развитие нельзя. Однако если своевременно обратиться к врачу, можно снизить тяжесть болезни и предотвратить развитие депрессии, аутоагрессии и обсессивно-компульсивного расстройства.
При сильных тиках, не поддающихся лечению и мешающих общаться, учиться или работать, пациент может пройти медико-социальную экспертизу: специалисты оценят тяжесть состояния и, при необходимости, определят группу инвалидности.
Синдром Байуотерса
Впервые этот синдром был выделен как отдельное заболевание в 1941 году английским врачом Эриком Байуотерсом, который лечил людей, пострадавших от бомбардировок в Лондоне во время Второй мировой войны [1]. У больных, которые длительное время провели под завалами со сдавленными конечностями, наблюдалась особая форма шока. Особенность заключалась в том, что при не слишком тяжелых повреждениях (внутренние органы у таких пациентов, как правило, не были травмированы) после комплекса лечебных мероприятий состояние больных существенно улучшалось, но затем наступало резкое ухудшение. У большинства пациентов развивалась острая почечная недостаточность и вскоре они умирали. Существует несколько вариантов названий этого синдрома: компартмент-синдром, компрессионная травма, краш-синдром (от англ. сrush - «раздавливание, смятие»), травматический токсикоз.
Байуотерсу удалось выявить три последовательные стадии, приводящие к развитию краш-синдрома:
- сдавливание конечности и последующий некроз тканей;
- развитие отека в месте сдавливания;
- развитие острой почечной недостаточности и ишемического токсикоза.
Патогенез
Синдром Байуотерса возникает в результате сдавливания конечности, повреждения основных сосудов и магистральных нервов. Подобная травма встречается примерно у 30% людей, пострадавших в результате природных или техногенных катастроф.
В патогенезе этого заболевания ведущее значение имеют три фактора: регуляторный, связанный с болевым воздействием на организм, существенная плазмопотеря и, наконец, тканевая токсемия. Отметим, что подобные факторы в той или иной степени наблюдаются практически при любой травме, но при краш-синдроме они проявляются особенно ярко. Каждый из этих факторов дает свой вклад в клиническую картину синдрома длительного сдавления.
Болевое воздействие влияет на человека, попавшего под завал, наиболее сильно. Отмечается рефлекторный спазм сосудов периферических органов и тканей, что приводит к нарушению газообмена и последующей гипоксии тканей. Сосудистый спазм и развивающаяся гипоксия вызывают дистрофические изменения эпителия почечных извитых канальцев, существенно падает клубочковая фильтрация.
Плазмопотеря развивается вскоре после травмы и даже после устранения причины сдавливания.
Плазмопотерю связывают с увеличением проницаемости капилляров на фоне травмы, что ведет к выходу плазмы крови из кровяного русла.
Объем циркулирующей крови уменьшается, вязкость возрастает, затрудняется транспорт кислорода. В месте повреждения развивается отек, многочисленные кровоизлияния, отток крови из сдавленной конечности нарушается, так как отечная жидкость приводит к сужению просвета кровеносных сосудов вплоть до их полной блокировки. В результате развивается ишемия конечности, в тканях усиленно накапливаются продукты клеточного метаболизма, нарастает количество миоглобина, креатинина, ионов калия и кальция. Увеличение концентрации миоглобина в циркулирующей крови, развивающийся метаболический ацидоз оказывают губительное влияние на работу почечных канальцев. Усугубляют токсемию и другие белковые факторы, которые накапливаются в результате сдавливания конечности и повреждения мышечной ткани. После восстановления кровообращения они «залпом» начинают поступать в сосудистое русло. В этот момент появляется ряд симптомов, характерных для ишемического токсикоза.
Интоксикация организма выражена тем сильнее, чем больше масса сдавленных тканей и длительность компрессионного воздействия.
Степени тяжести краш-синдрома
В зависимости от объёма повреждения и длительности сдавления, выделяют 4 степени тяжести синдрома [2].
Легкая степень - сдавление небольшого сегмента конечности в течение не более чем двух часов. В этом случае токсемия выражена слабо, хотя отмечаются острая почечная недостаточность и нарушения гемодинамики. В большинстве случаев при проведении своевременной терапии улучшение наступает в течение недели.
Средняя степень возникает при сдавлении конечности целиком в течение четырех часов. Подобное состояние характеризуется интоксикацией, миоглобинурией и олигоурией.
Длительная компрессия конечностей (4-7 часов) ведет к проявлению симптомов, характерных для тяжелой степени синдрома Байуотерса. Отмечаются существенные нарушения гемодинамики, выражены симптомы интоксикации, быстро развивается острая почечная недостаточность.
Несвоевременное и неправильное оказание медицинской помощи в большинстве случаев ведет к летальному исходу.
Также важно правильно и быстро действовать, если у пациента диагностирована крайне тяжелая степень краш-синдрома. Такой диагноз ставят при сдавлении нижних конечностей в течение 8 и более часов. Развивающийся ишемический токсикоз окажется губительным для пациента вскоре после проведенной декомпрессии. Смертность таких больных крайне высока даже при проведении своевременного лечения.
Выбор подхода к лечению начинают с оценки степени компрессии и длительности сдавления конечностей. Для специалистов, принимающих участие в спасательных операциях, важно постараться освободить максимальное число пострадавших в первые два часа после возникновения чрезвычайной ситуации. Именно в этом случае прогноз окажется благоприятным для большинства пациентов.
Во время землетрясения в Мармаре (Турция), произошедшего в 1999 году, пострадало много детей. Тогда был накоплен колоссальный опыт по устранению последствий компрессионной травмы у маленьких пациентов. Специфика лечения синдрома Байуотерса у детей обусловлена тем, что их травмы зачастую оказываются гораздо тяжелее, чем у взрослых [3].
С детьми сложнее коммуницировать в процессе спасательной операции, поэтому часто они проводят под завалами больше времени, чем взрослые. Детский организм в большей степени подвержен переохлаждению и потере жидкости, так что следует особое внимание уделить регидратации сразу же после спасения ребенка.
Вне зависимости от степени тяжести и возраста пациента проводят противошоковые мероприятия: вводят анальгетики, сердечно-сосудистые препараты для нормализации артериального давления. В большинстве случаев это делают еще до извлечения пострадавшего из-под завала.
Лечение, начатое еще до удаления пресса, дает возможность избежать развития ишемического токсикоза. В первую очередь это касается обширных компрессионных травм.
После освобождения поврежденной конечности на место сдавливания накладывают жгут, что помогает не допустить «залпового» выброса накопленных токсических веществ в кровяное русло. Это важная особенность оказания медицинской помощи при синдроме Байуотерса. После перемещения пострадавшего и устранения сдавления конечность бинтуют с помощью эластичного бинта, и только тогда удаляют жгут. Также рекомендовано охлаждение поврежденной конечности.
Соблюдение последовательности этапов лечения пациентов с компрессионными травмами очень важно. Своевременное использование инфузионной терапии, понимание патогенеза синдрома Байоутерса существенно увеличивает количество спасенных жизней.
При легкой степени синдрома хирургического лечения не проводят, нередко такие больные лечатся амбулаторно. При средней степени тяжести нарушения гемодинамики выражены достаточно ярко: отек нарастает, нарушается микроциркуляция, увеличивается количество микротромбозов, однако хирургическое лечение и в этом случае показано не всегда. Рекомендована инфузионная терапия, которая позволяет не допустить развития или же прогрессирования острой почечной недостаточности.
В случаях тяжелой и крайне тяжелой степени выраженности краш-синдрома консервативное лечение неэффективно, и необходимо хирургическое лечение. Проводят фасциотомию поврежденной конечности, которая способствует восстановлению кровообращения и дает возможность избежать полной некротизации конечности. Нередко приходится ампутировать дистальные отделы конечностей, чтобы спасти пациента.
Параллельно проводится терапия острой почечной недостаточности - назначается строгий питьевой режим, гемодиализ, плазмоферез и инфузионная терапия (введение растворов глюкозы, альбумина и т.п.).
В реабилитационном периоде следует уделять внимание физиопроцедурам (например, массажу) и лечебной физкультуре, которые способствуют более эффективному восстановлению конечности, минимизируя атрофию мышц и нервов.
Случай из практики
В результате автомобильной катастрофы 21-летний молодой мужчина провел 10 часов, будучи зажатым в поврежденном автомобиле. Он был доставлен в больницу города Низва (Оман), находясь в полном сознании [4]. Осмотр показал, что грудная клетка, брюшная полость, спина и таз не были повреждены. В то же время наблюдался отек правого плеча, правая верхняя конечность была обездвижена. Рентгеновское исследование выявило перелом правой ключицы.
Также отмечался отек правой нижней конечности, кожный покров поврежден не был. На левой ноге был диффузный отек, затрагивавший голень и бедро, а также глубокие ссадины. Обе ноги были практически неподвижны в голеностопных суставах, отмечались нарушения чувствительности в области голеней. Допплерографическое исследование показало нарушение венозного кровотока в стопе и голени. Дальнейшее наблюдение выявило быстрое накопление креатинина, миоглобина, калия в сыворотке крови, а также миоглобинурию.
Проводилась инфузионная терапия: физиологический раствор, глюкоза, бикарбонат натрия. Несмотря на это у пациента развилась анурия, а уровень калия в крови продолжал повышаться. Пострадавшему назначили гемодиализ и провели фасциотомию левого бедра и голени, в результате которой обнаружили, что часть бедренных мышц некротизирована. На 7-й день лечения в мазке из раны были обнаружены грамотрицательные бактерии - E.coli и бактерии рода Proteus. Пациенту назначили адекватную антибиотикотерапию, рана регулярно обрабатывалась антисептиками. Состояние пациента прогрессивно ухудшалось. Несмотря на прием антибиотиков, развилась бактериальная септицемия, в связи с чем была рекомендована ампутация левой ноги, от которой пациент и его семья отказались. Ими было принято решение продолжить лечение за границей, где пострадавший скончался от тяжелого сепсиса через три дня после прибытия.
Резюме
Синдром Байуотерса был выделен как нозологическая единица не так давно - лишь в середине 20 века. При спасении и последующем лечении пострадавших с тяжелыми компрессионными травмами важны координированные действия спасателей и врачей. Быстрое извлечение людей из-под завалов и оказание первой помощи еще до удаления пресса минимизирует тяжелые последствия синдрома длительного сдавления конечностей и помогает сохранить жизнь больного.
1.Bywaters EG. Crushing Injury. Br Med J. 1942 Nov 28; Vol.2 No.4273. P.643-6.
2.Рудаев В.И. Кричевский А.Л., Галеев И.К. Краш-синдром в условиях катастроф. - Методические рекомендации для реанимационно-противошоковых групп ВГСЧ, специализированных бригад постоянной готовности Службы медицины катастроф и реанимационных бригад скорой медицинской помощи. 1999.
3.Dario Gonzalez. Crush syndrome. Crit Care Med. 2005. Vol. 33, No. 1 (Suppl.). S.34-41.
4.Dinesh Dhar, TP Varghese. Crush Syndrome Case Report and Literature Review. Macedonian Journal of Medical Sciences. 2010 Sep 15; 3(3):319-323.
Синдром Барттера
Синдром Барттера - это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.

Общие сведения
Синдром Барттера в клинической урологии представляет собой редкую генную мутацию - дефект петли Генле, наследуемую по аутосомно-рецессивному типу и проявляющуюся, как правило, уже в детском возрасте. Неспособность почечных нефронов задерживать калий приводит к хронической потере его с мочой и уменьшению объема циркулирующей крови при нормальном или пониженном АД. В зависимости от вида пораженных генов различают: неонатальный синдром Барттера 1 и 2 типов, классический синдром Барттера, синдром Гительмана. Также встречается приобретенный синдром псевдо-Барттера, характеризующийся сходными проявлениями, но не сопровождающийся патологией почечных канальцев.
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике - сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Читайте также:
- Уровень физической нагрузки при алкоголизме. Влияние алкоголя на выносливость
- Индукция иммунного ответа. Регуляция иммунного ответа
- Иммуноглобулин Е при вирусных инфекциях.
- Апоневротические швы. Техника наложения швов на апоневроз.
- Гипоталамический контроль гипофиза. Гипоталамо-гипофизарные кровеносные сосуды