Синдром Бушке-Оллендорф (Buschke-Ollendorff) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 01.02.2026
Синдром Бушке — Оллендорф (s. Buschke — Ollendorf), диссеминированный дерматофиброз с остеопойкилией [1] . Впервые описан немецким дерматологом Авраамом Бушке и американским врачом Элен Оллендорф Курт [2] в 1928 году Синдром Бушке — Оллендорфа наследуется по аутосомно-доминантному типу [3] .
Семейно-наследственная конституциональная аномалия среднего зародышевого листка. На коже в верхнем отделе спины, живота, затылка, плечах, бедрах, пояснично-крестцовой области появляется сыпь, расположенная симметрично, светло-жёлтоватого цвета, плотной консистенции, слегка поднимающаяся над уровнем кожи.
Рентгенологически на фоне почти нормальной кости в губчатом веществе эпифизов и метафизов длинных трубчатых костей, плюсневых и пястных костей, фаланг наблюдаются пятнистые очаги затемнения. Реже поражаются позвонки, рёбра, грудина, череп. Иногда заболевание сочетается с синдромом синих склер, несовершенным костеобразованием, дебильностью, эпилепсией.
Синдром Бушке-Оллендорфа
Синдром Бушке-Оллендорфа наследственное заболевание соединительной ткани. Редкое заболевание поражает скелет и кожу. Как влияет синдром Бушке-Оллендорфа на организм человека и как лечить это заболевание?
Что такое синдром Бушке-Оллендорфа?
Синдром Бушке-Оллендорфа, который также известен под латинским названием дерматофиброз лентикулярного распространения, был назван в честь немецкого дерматолога Абрахама Бушке и немецко-американского дерматолога Хелен Оллендорф-Курт. Редкое заболевание соединительной ткани представляет собой сочетание изменения кожи и изменения скелета. Типичные симптомы включают остеопоицилоз или низкий рост.
причины
Это очень редкое заболевание является генетическим и поэтому не полностью излечимо. Причина этого дефекта - мутация хромосомы в гене LEMD3. В этом гене происходит так называемая мутация потери функции. Считается, что это генная мутация, встречающаяся в генетике. Следствием этого является потеря функции соответствующего генного продукта.
При синдроме Бушке-Оллендорфа соответствующему гену предписывается производить белок. Сигнальные пути регулируют различные клеточные процессы и рост клеток. Это также включает рост новых костных клеток. Мутация гена LEMD3 снижает количество функциональных белков. Помимо прочего, это может увеличить плотность кости. Нет четких доказательств этого исследования.
Симптомы, недуги и признаки
Первые симптомы могут появиться в любой возрастной категории. Первые признаки часто можно распознать в детстве, при этом симптомы проявляются по-разному как в костях, так и на коже. В большинстве случаев изменения кожи безболезненны и со временем разрастаются. Эта особенность проявляется через некоторое время после рождения ребенка.
Эти изменения в соединительной ткани имеют размер всего несколько миллиметров и в основном расположены на руках и ногах, а также на туловище. Остеопойкилоз - это доброкачественный и случайно обнаруженный порок развития кости. Этот порок развития кости редко диагностируется в зрелом возрасте. В кости видны небольшие круглые участки, указывающие на повышенную плотность кости.
Это видно на рентгеновском снимке в виде светлых пятен. Пороки развития и кожные изменения обычно не решают никаких жалоб, поэтому диагноз очень трудно подтвердить. Синдром Бушке-Оллендорфа в большинстве случаев диагностируется случайно.
Диагностика и курс
Чтобы правильно диагностировать синдром Бушке-Оллендорфа, помимо генетического теста проводятся дополнительные обследования. При этом заболевании человеческого организма простого лабораторного исследования недостаточно. Изменения кожных покровов подтверждены гистологическим исследованием. Поражения кожи исследуют под микроскопом путем окрашивания срезов ткани.
Этот метод диагностики можно использовать для выявления аномалий соединительной ткани разной степени. Особенно страдают эластичные волокна и волокна коллагена. Они отвечают за прочность ткани на разрыв. Кожные изменения находятся на небольшом пространстве и близко друг к другу. Цвет от беловатого до желтого, форма овальная. Размер между горошиной и чечевицей.
Для определения изменений в скелете делают рентгеновские снимки кистей и стоп. Поскольку поражения костей в детстве в основном не замечаются, эти характеристики синдрома Бушке-Оллендорфа обнаруживаются только в пожилом возрасте. Типичные изменения костей не наблюдаются до 15 лет.
На рентгенограмме видны увеличенные и слегка утолщенные трабекулы, то есть небольшие трабекулы на кости. Эти аномалии кости не следует путать с метастазами в кости или мелореостозом (утолщением кости). Если возникает боль, синдром Бушке-Оллендорфа возникает не сразу.
Чтобы можно было включить это наследственное заболевание в диагноз, необходимо объединить несколько жалоб пациента. Если люди страдают отеками суставов или частыми излияниями в суставы, это не означает, что обследования выявляют синдром Бушке-Оллендорфа.
осложнения
Во многих случаях синдром Бушке-Оллендорфа диагностируется поздно, поэтому лечение может быть отложено. Это связано с тем, что больные обычно не испытывают боли или другого дискомфорта от синдрома. Могут возникать как злокачественные, так и доброкачественные пороки развития костей.
Как правило, сам диагноз ставится случайно при осмотре. Во многих случаях также наблюдаются отеки или выпот на суставах. У пациента обычно больше нет жалоб или осложнений. По этой причине лечение синдрома Бушке-Оллендорфа необходимо не во всех случаях.
Если на коже появятся какие-либо видимые аномалии, эти участки можно обработать. Чаще всего это делается с помощью лекарств или мазей. Это всегда приводит к положительному течению болезни без дальнейших жалоб и осложнений.
Диагностика часто затруднена, поскольку влияние синдрома Бушке-Оллендорфа на кости можно увидеть только в небольшой степени на рентгеновских снимках. Чаще всего страдают только молодые люди в возрасте 15 лет и старше. На продолжительность жизни не влияет синдром Бушке-Оллендорфа.
Когда нужно идти к врачу?
В большинстве случаев синдром Бушке-Оллендорфа диагностируется в детстве. Затем следует проконсультироваться с врачом, если у пациента наблюдаются различные кожные изменения. Хотя они не связаны с болью, их всегда должен осматривать врач. Это может предотвратить дальнейшие осложнения. Рост этих кожных изменений также может указывать на синдром. По этой причине кости пациента также следует исследовать на предмет изменений.
Сама плотность костной ткани повышена, что можно определить с помощью рентгена. Ранняя диагностика важна, даже если у детей нет других симптомов или ограничений. Диагноз может поставить терапевт или педиатр. Дальнейшее лечение обычно проводится с помощью мазей или таблеток, поэтому специального врача не требуется. Чем раньше диагностируется заболевание, тем лучше можно ограничить симптомы и лечить.
Врачи и терапевты в вашем районе
Лечение и терапия
Чтобы распознать синдром Бушке-Оллендорфа как таковой, большое значение также имеет семейный анамнез пациента. Не все пациенты имеют одинаковые симптомы. При кожных аномалиях не обязательно наличие аномалии костей, и наоборот.
В целом синдром Бушке-Оллендорфа не вызывает никаких симптомов. Однако, чтобы избежать неправильного лечения симптомов, необходима подробная диагностика заболевания. Чем раньше будет правильно диагностировано заболевание, тем раньше можно будет начать соответствующую терапию. При необходимости кожные аномалии лечат симптоматически на пораженном участке кожи.
Лечение можно проводить мазями или таблетками. Правильный метод лечения индивидуально адаптирован к пациенту и тяжести жалоб. Соответствующие лекарства назначаются при тяжелых скелетных симптомах. Однако не известно ни одного случая, который принимал бы такие серьезные масштабы.
Прогноз и прогноз
Синдром Бушке-Оллендорфа нельзя лечить причинно. Лечить можно только отдельные симптомы, а полного излечения не бывает.
Как правило, пораженные страдают различными пороками развития, которые могут быть разной степени тяжести. От этих пороков развития также зависит дальнейшее течение и лечение заболевания, поскольку для их облегчения обычно также необходимы хирургические вмешательства. Симптоматически лечат только пораженные участки кожи. Никаких сложностей.
Чем раньше начнется лечение синдрома, тем выше шансы на положительное течение болезни. На данный момент не известно серьезных случаев синдрома Бушке-Оллендорфа, при которых ожидаемая продолжительность жизни пострадавшего была сокращена. Хотя пациенты зависят от постоянного лечения и использования мазей и лекарств в своей жизни, их повседневная жизнь не ограничена или ограничена лишь незначительно.
профилактика
Поскольку синдром Бушке-Оллендорфа - заболевание, которое сложно распознать, правильно назначить симптомы непросто. Поскольку это генетически наследственное заболевание, с ним невозможно бороться. Вероятность того, что болезнь находится в гене, очень мала.
В мире около 20 000 рождений, и в среднем только один человек страдает синдромом Бушке-Оллендорфа. Даже если диагностировано наследственное заболевание, оно не влияет на продолжительность жизни. У пациентов хороший прогноз здоровья, и современная медицина может полностью сдержать любые симптомы, которые могут возникнуть. При первых признаках заболевания необходима обширная диагностика.
уход за выздоравливающим
Как правило, пациенты с синдромом Бушке-Оллендорфа не имеют прямых возможностей для последующего наблюдения. Заболевание можно лечить только симптоматически, а не причинно, поэтому больной в основном зависит от пожизненной терапии. Это также может привести к сокращению продолжительности жизни.
Поскольку синдром Бушке-Оллендорфа обычно лечится с помощью таблеток, кремов или мазей, пострадавший должен обеспечить их регулярный прием или применение. Также следует рассмотреть возможные взаимодействия с другими лекарствами и обсудить их с врачом.
Как правило, симптомы не сильно ограничивают пострадавшего, поэтому пациент может жить обычной жизнью. Точно так же не известны особо тяжелые формы синдрома Бушке-Оллендорфа. Поскольку синдром Бушке-Оллендорфа в некоторых случаях может привести к психологическим жалобам или депрессивным настроениям, в этих случаях следует посетить психолога.
Однако разговор с друзьями или семьей также может помочь избежать такого дискомфорта. Кроме того, в случае синдрома Бушке-Оллендорфа также полезен контакт с другими пациентами, страдающими этим синдромом, поскольку это может привести к обмену информацией.
Ты можешь сделать это сам
Синдром Бушке-Оллендорфа теперь можно хорошо лечить. Пациент может дополнительно поддержать выздоровление, соблюдая строгую личную гигиену и ухаживая за пораженными участками в соответствии с указаниями врача.
Также важно использовать предписанные средства по уходу. Лечебные мази и лосьоны необходимо наносить регулярно, особенно в области изменений кожи, чтобы ускорить заживление ран и предотвратить образование рубцов. Пациенту также следует избегать воздействия солнечных лучей и контакта кожи с вредными веществами. Этого можно достичь, например, надев подходящую одежду. Людям, которые подвергают свою кожу сильному стрессу на работе, следует подумать о смене работы.
Кроме того, регулярные проверки являются частью необходимой самопомощи. Любой, кто замечает необычные места в других частях тела или страдает аллергией на назначенные средства, должен сообщить об этом своему врачу. Врач часто может дать дополнительные советы о том, как пациент может поддержать лечение. В первую очередь важны отдых и отдых, чтобы организм мог в достаточной мере восстановиться. В зависимости от причины синдрома могут быть полезны дальнейшие меры, которые пострадавшие могут обсудить со своим семейным врачом.
Синдром Бушке-Оллендорф (Buschke-Ollendorff) - синонимы, авторы, клиника
Российский государственный медицинский университет им. Н.И. Пирогова
ФГБОУ ВО «Российский национальный исследовательский университет им. Н.И. Пирогова», Москва, Россия;
Российская детская клиническая больница, Москва, Россия
Сложный диагностический случай: синдром Бушке—Оллендорф или соединительнотканный невус?
Дисплазия соединительной ткани, представляющая собой генетически детерминированное нарушение закладки и постнатального развития, является весомой проблемой современной медицины в силу широкого распространения и возможных тяжелых последствий. Клинические проявления дисплазии соединительной ткани варьируют в широких пределах — от повышенной растяжимости кожи до сосудистых аномалий, приводящих к внезапной смерти. Большую группу в пределах дисплазии соединительной ткани составляют различные невусы, изолированные или в составе различных синдромов. Одним из редких генетически детерминированных синдромов является сидром Бушке—Оллендорф, относящийся к орфанным заболеваниям с аутосомно-доминантным наследованием. Основные клинические проявления синдрома — распространенные соединительнотканные невусы кожи, выявляемые в раннем детском возрасте, и остеопойкилия, чаще диагностируемая у взрослых. К редким проявлениям синдрома Бушке—Оллендорф относят различные пороки и поражения нервной системы (от нарушения когнитивного развития до эпилепсии). Диагностика синдрома основывается на совокупности определенных симптомов, главными из которых являются соединительнотканные невусы и остеопойкилия. В случае неполного синдрома диагностика базируется на молекулярно-генетическом исследовании, которое в силу дороговизны доступно не всем пациентам. Данный клинический случай демонстрирует трудность диагностического поиска и неоднозначность получаемых при этом результатов. Представленный клинический случай был обсужден на общегородском Московском консилиуме при участии дерматовенерологов Москвы и профессоров Н.Н. Потекаева, В.Г. Акимова, В.Н. Гребенюка, А.Н. Львова, Э.А. Баткаева, Н.Г. Короткого, О.В. Жуковой, В.А. Волнухина.
Дисплазия соединительной ткани — это нарушение развития соединительной ткани в эмбриональном и постнатальном периодах, генетически детерминированное, характеризующееся дефектами волокнистых структур и основного вещества соединительной ткани, приводящее к расстройству гомеостаза на тканевом, органном и организменном уровнях в виде различных морфофункциональных нарушений висцеральных и локомоторных органов с прогредиентным течением, определяющее особенности ассоциированной патологии. По самым скромным данным, показатели распространенности данной группы заболеваний соотносятся с распространенностью основных социально значимых неинфекционных заболеваний. К данной группе заболеваний относится диссеминированный лентикулярный дерматофиброз (синдром Бушке—Оллендорф), являющийся редким наследственным заболеванием [1—3]. Впервые заболевание было описано Абрахамом Бушке в 1902 г. под названием «scleroderma adultorum». В 1915 г. Генрих Эрнст Альберс-Шёнберг, а затем в 1928 г. Элен Оллендорф описали еще 2 случая заболевания.
Синдром Бушке—Оллендорф представляет собой сочетание соединительнотканных невусов кожи и остеопойкилии (OMIM 166700). Частота встречаемости данного заболевания составляет 1 на 20—30 тыс. живых новорожденных, с одинаковой частотой у мальчиков и девочек. Заболевание наследуется аутосомно-доминантно с высокой пенетрантностью и вариабельной экспрессивностью. В основе лежит мутация гена LEMD3, расположенного на 12q14.3 хромосоме, кодирующего один из структурных белков соединительной ткани, что сопровождается чрезмерным накоплением эластина в дерме [4].
Синдром Бушке—Оллендорф обычно проявляется рано, возможна манифестация первых симптомов сразу после рождения. Как и при любом пороке развития, симптоматика весьма разнообразна и подразделяется на кожные и внекожные признаки (рис. 1).

Рис. 1. Клинические проявления синдрома Бушке—Оллендорф.
Первым симптомом заболевания является поражение кожного покрова в виде соединительнотканного невуса. Высыпания обычно локализуются на боковых поверхностях туловища, верхней трети живота, спины, пояснице, бедрах, ягодицах; представлены папулами, узлами и бляшками, слегка возвышающимися над уровнем кожи, телесного или желтоватого цвета, склонными к сетевидной, линейной или герпетиформной ориентации; могут сливаться между собой. Элементы сыпи, как правило, симметричны, однако в детском возрасте описано одностороннее расположение. Консистенция элементов мягкая, пальпация безболезненна. Субъективных ощущений от эффлоресценций обычно нет. Из других кожных изменений описаны анетодермия, ладонно-подошвенный кератоз, морфея, околосуставные подушечки.
Вторым по частоте встречаемости признаком является поражение костной системы в виде остеопойкилии (синонимы — врожденная рассеянная склерозирующая остеопатия, врожденная пятнистая множественная остеопатия), возникающей в результате очагового отложения кальция [3, 5]. Признаки остеопойкилии обнаруживаются преимущественно в костях конечностей и плечевого пояса. Клинически они не вызывают жалоб, но важны для дифференциальной диагностики. При рентгенологическом исследовании в костях выявляются мелкие округлые или овальные уплотнения спонгиозных структур костной ткани (рис. 2).
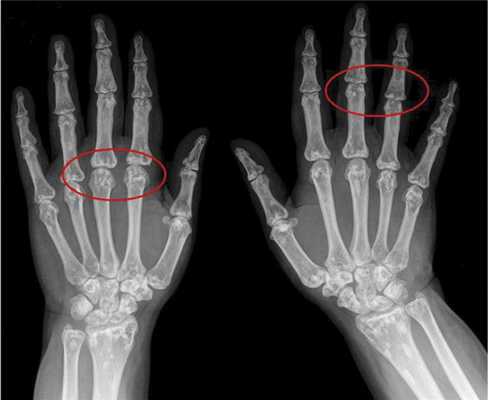
Рис. 2. Остеопойкилия.
Более редкими проявлениями заболевания являются различные пороки развития сердца, ребер, зубов, лицевого скелета, глаз и т. д., эндокринные расстройства (сахарный диабет), неврологические нарушения (эпилепсия, умственная отсталость).
Диагностика синдрома опирается на сочетание типичных клинических симптомов и требует обязательного инструментального подтверждения — морфологического, рентгенологического, генетического. При гистологическом исследовании выявляют признаки соединительнотканного невуса: локальную пролиферацию эластических структур и накопление муцина в дерме. Для подтверждения синдрома Бушке—Оллендорф необходимы дополнительные методы окраски гистопрепаратов по Вейгерту, по Ван-Гизону, толуидиновым синим и сравнение с биоптатом здоровой кожи (рис. 3).
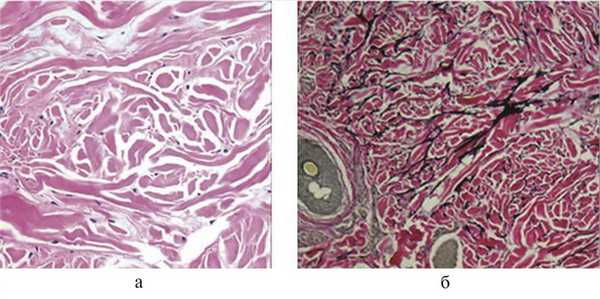
Рис. 3. Морфологическое исследование биоптата (кожа передней брюшной стенки) пациента с синдромом Бушке—Оллендорф. а — участки уплотнения коллагеновых пучков без признаков склерозирования (окраска гематоксилином и эозином; ×40); б — утолщенные пучки эластических волокон, ориентированные в различных направлениях (окраска по Ван-Гизону, ×40).
При невозможности постановки диагноза с помощью рутинных методов применяют молекулярно-генетическую диагностику, заключающуюся в поиске мутаций в гене LEMD3. Течение заболевания в большинстве случаев доброкачественное, хотя возможны и неблагоприятные варианты, приводящие к развитию злокачественных новообразований в области остеопойкилии.
Демонстрацией нашего клинического случая мы хотим показать сложность диагностики и дифференциальной диагностики генодерматозов.
Клинический случай
Пациент А., 5 лет. Ребенок от 6-й беременности (1-я — неразвивающаяся, 2-я и 5-я — доношенный мальчик, 3-я - медицинский аборт, 4-я — выкидыш), протекавшей на фоне угрозы прерывания, анемии, ОРВИ. Роды (третьи) путем экстренного кесарева сечения на 36-й неделе (несостоятельность рубца на матке). Масса тела при рождении 2630 г, рост 48 см, массо-ростовой показатель 55 (норма 60—80), оценка по шкале Апгар 7—8 баллов. Раннее развитие без особенностей. Грудное вскармливание до 18 мес. Вакцинация в соответствии с Национальным календарем прививок. Генеалогический анамнез отягощен по линии матери — аллергические заболевания (атопический дерматит, поллиноз), феномен Вольфа—Паркинсона—Уайта (WPW), артропатии. У старшего сибса пауциартикулярный юношеский артрит, кольцевидная гранулема; у младшего — поллиноз, пищевая аллергия.
Мама считает ребенка больным с рождения, когда после выписки из родильного дома на передней поверхности правого бедра было обнаружено розовое пятно размером до 1 см, округлой формы с четкими контурами. Педиатр на первом патронаже расценил высыпания как проявления аллергической реакции на пищу, рекомендовал маме придерживаться диеты. В возрасте 2—3 мес у ребенка появились эритемато-сквамозные высыпания в области лица (на щеке), туловища, голеней, возникающие на фоне погрешности в питании матери. Элемент на бедре по-прежнему расценивался как проявление пищевой аллергии. Впервые по поводу высыпаний обратились к аллергологу в возрасте 6 мес, был выставлен диагноз атопического дерматита и рекомендована гипоаллергенная диета.
Прогрессирование высыпаний началось после 1,5 лет, когда после перенесенной ОРВИ начали появляться новые элементы, изменился их внешний вид (приобрели желтоватый оттенок) и консистенция (стали возвышаться над поверхностью кожи и уплотнились). Аллерголог по месту жительства назначил терапию топическими глюкокортикостероидами (адвантан) и антигистаминным препаратом 1-го поколения диметинденом (фенистил). Эффекта от терапии не отмечалось. Резкое увеличение количества высыпных элементов отметили в возрасте 3 лет после перенесенной катаральной ангины, а затем через 5 мес после кишечной инфекции (рис. 4).
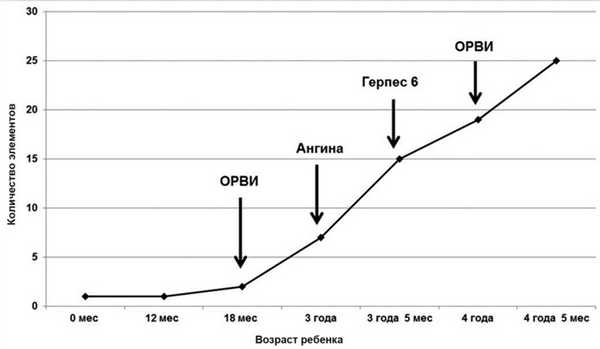
Рис. 4. Динамика кожного процесса. С этого же времени ребенок впервые стал жаловаться на боли в ногах.
За полуторагодовалый период ребенок неоднократно консультирован различными специалистами, проходил сложные и дорогостоящие исследования (табл. 1, 2).
Таблица 2. Проведенные исследования Таблица 1. Консультации специалистов
При осмотре в отделении состояние ребенка удовлетворительное, самочувствие не нарушено. Патологический кожный процесс невоспалительного характера, ограниченный, асимметричный; локализован по передней, боковой и задней поверхностям правого бедра. Представлен подкожными узлами от 0,5 до 1,5 см, желтовато-розового цвета с четкими контурами, полушаровидной формы, с гладкой поверхностью, плотноэластической консистенции, склонными к слиянию (рис. 6).
Рис. 6. Кожный процесс у ребенка А., 5 лет. Задняя и боковая поверхности правого бедра. Пальпация элементов безболезненная; субъективно — зуд после контакта с источником тепла. Кожа вне очагов не изменена, умеренно выражен ксероз. Придатки кожи не изменены, слизистые оболочки интактны. Рис. 5. Рентгенологическое исследование у больного А., 5 лет. а — кисти; б — стопы; в — коленные и голеностопные суставы.
При рутинном обследовании, включавшем клинический анализ крови, общий анализ мочи, биохимическое исследование крови, иммунный статус, патологии не выявлено. При УЗ-исследовании брюшной полости и забрюшинного пространства выявлена ротация обеих почек. Невропатолог диагностировал синдром гиперактивности, аллерголог — пищевую аллергию. На момент осмотра данных, свидетельствующих о синдроме Бушке—Оллендорф, не получено. Заключительный дерматологический диагноз: соединительнотканный невус.
Рекомендации заключались в динамическом наблюдении, фотодокументировании и проведении УЗ-исследования эффлоресценций в области правого бедра 1 раз в 6 мес. По достижении ребенком совершеннолетия возможна хирургическая коррекция, заключающаяся в иссечении невоидных образований. Динамическое наблюдение необходимо, так как возможно отсроченное появление других клинических признаков, входящих в состав синдрома Бушке—Оллендорф, прогноз для выздоровления при этом неблагоприятный. Основной причиной смерти данных пациентов являются онкологические процессы (остеосаркома, хондросаркома), пороки сердца и осложненный сахарный диабет. Своевременная диагностика, грамотная дифференциальная диагностика и правильное ведение таких пациентов необходимо для профилактики осложнений.
Несмотря на схожесть клинических проявлений у ребенка с синдромом Бушке—Оллендорф (типичные кожные проявления), отсутствие типичных поломок в гене LEMD3 позволяет полностью исключить синдром Бушке—Оллендорф. Для определения прогноза необходимо продолжить диагностический поиск (иммуногистохимическое исследование соединительнотканного невуса, полноэкзомное секвенирование фибробластов кожи), что позволит дифференцировать относительно доброкачественные заболевания — множественные дермальные невусы коллагенового или эластинового типов, тяжелого мелореостоза, инвалидизирующего с возрастом.
Синдром Бушке - Оллендорфа - Buschke-Ollendorff syndrome

Синдром Бушке - Оллендорфа - редкое генетическое заболевание связан с LEMD3. Считается, что он наследуется аутосомным доминантным способом. Он назван в честь Авраама Бушке и Хелен Оллендорф Курт, которые описали его у 45-летней женщины. Его частота составляет почти 1 случай на каждые 20000 человек, и он одинаково встречается как у мужчин, так и у женщин.
Содержание
- 1 Признаки и симптомы
- 2 Патогенез
- 3 Диагноз
- 3.1 Дифференциальный диагноз
Признаки и симптомы
Признаки и симптомы этого состояния соответствуют следующие (возможные осложнения включают стеноз аорты и потеря слуха ):
Патогенез
Синдром Бушке - Оллендорфа вызван одним важным фактором: мутациями в гене LEMD3 (12q14), локализованном на хромосоме 12.
Среди С генетической точки зрения важными аспектами синдрома Бушке-Оллендорфа являются:
- LEMD3 (белок), также называемый MAN1, является важным белком во внутренней ядерной мембране.
- LEMD3 ген g Содержит инструкции по производству белка, который контролирует передачу сигналов для трансформирующего фактора роста-бета.
- ген LEMD3 помогает в костном морфогенном белке пути
- Оба вышеупомянутых пути помогают выращивать новые костные клетки
- пути BMP и TGF-β контролируют белки SMAD, которые затем связываются с ДНК
- LEMD3 после мутации, вызывая уменьшение белка, что, в свою очередь, вызывает избыток
Диагноз
Гистопатология дерматофиброза рассеянного лентикулярного.
Диагноз этого состояния может быть установлен с помощью нескольких методов, одним из таких методов является генетическое тестирование, а также:
Дифференциальный диагноз
Дифференциальный диагноз для человека, предположительно страдающего синдромом Бушке - Оллендорфа, следующий:
Лечение
Что касается лечения Буша Синдром Ке - Оллендорфа, при возникновении осложнения стеноза аорты может потребоваться операция. Для лечения потери слуха также может потребоваться хирургическое вмешательство.
Синдром Бушке - Оллендорфа редкий генетическое расстройство связан с LEMD3. Считается, что он передается по наследству аутосомный доминирующий манера. [5] Он назван в честь Авраам Бушке и Хелен Оллендорф Курт, [6] который описал это у 45-летней женщины. Его частота составляет почти 1 случай на каждые 20 000 человек, и он в равной степени встречается как у мужчин, так и у женщин. [4]
Признаки и симптомы этого состояния соответствуют следующему (возможные осложнения включают: стеноз аорты и потеря слуха [2] [4] ):
Синдром Бушке - Оллендорфа вызван одним важным фактором: мутациями в гене LEMD3 (12q14), расположенном на хромосома 12. [ нужна цитата ]
Среди важных аспектов состояния синдрома Бушке - Оллендорфа, генетически говоря, являются: [7] [8] [9]
- LEMD3 (белок), также называемый MAN1, является важным белком в внутренняя ядерная мембрана.
- Ген LEMD3 дает инструкции по производству белка, который контролирует передачу сигналов для трансформирующий фактор роста-бета.
- Ген LEMD3 помогает в костный морфогенный белок путь
- Оба вышеуказанных пути помогают выращивать новые костные клетки
- Контроль путей BMP и TGF-β SMAD белки, которые затем связываются с ДНК
- После мутации LEMD3 вызывает сокращение белка, что, в свою очередь, вызывает избыток двух вышеуказанных путей.
Диагностика
![]()
Диагноз этого состояния можно установить с помощью нескольких методов, одним из таких методов является генетическое тестирование, а также: [2] [3]
Дифференциальный диагноз для человека с синдромом Бушке-Оллендорфа заключается в следующем: [3]
Что касается лечения синдрома Бушке - Оллендорфа, если осложнение: стеноз аорты происходит, тогда может потребоваться операция. Лечение потери слуха также может потребовать хирургического вмешательства. [4]
Читайте также:
- Бактерицидные антибиотики. Постантибиотический эффект (ПАЭ)
- ОНМК. Нарушения артериального кровообращения головного мозга. Острая гипертензивная энцефалопатия.
- Безоперационные средства увеличения груди: реальность или фикция?
- Иннервация гортани. Участие нервов гортани в формировании голоса
- Случай успешного удаления вросшего ногтя при нечувствительности к анестезии
