Синдром Цапперта (Zappert) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 22.01.2026
Что такое Синдром Геллера (симбиотический психоз, детская деменция, болезнь Геллера - Цапперта) -
Быстро прогрессирующее слабоумие у детей раннего возраста (после периода нормального развития) с отчетливой потерей на протяжении нескольких месяцев ранее приобретенных навыков, с появлением аномалий социального, коммуникативного или поведенческого функционирования.
Что провоцирует / Причины Синдрома Геллера (симбиотического психоза, детской деменции, болезни Геллера - Цапперта):
Причины расстройства не выяснены. Преобладает представление о наличии органической природы заболевания.
Симптомы Синдрома Геллера (симбиотического психоза, детской деменции, болезни Геллера - Цапперта):
После периода нормального развития до 2-3 лет в течение 6- 12 мес. формируется тотальное слабоумие. Часто отмечается продромальный период неясной болезни: ребенок становится своенравным, раздражительным, тревожным и гиперактивным. Речь обедняется, а затем распадается. Утрачиваются ранее приобретенные поведенческие, игровые и социальные навыки. Теряется контроль за функцией кишечника и мочевого пузыря. Интерес к окружающей обстановке потерян, характерны стереотипные двигательные действия. За ухудшением в течение нескольких месяцев следует состояние плато, затем может наступить незначительное улучшение. Расстройство часто сочетается с прогрессирующим неврологическим состоянием, которое обычно кодируется отдельно.
Прогноз заболевания неблагоприятный. Большинство больных остаются с тяжелой умственной отсталостью.
Диагностика Синдрома Геллера (симбиотического психоза, детской деменции, болезни Геллера - Цапперта):
Расстройство напоминает дементные состояния взрослого возраста, но отличается в 3 аспектах: 1) нет доказательств распознанного органического заболевания или повреждения; 2) потеря приобретенных навыков может сопровождаться некоторой степенью выздоровления и восстановления функций; 3) нарушения общения имеют характер, сходный с аутизмом, а не с интеллектуальным снижением.
Дифференциальная диагностика
Проводится с аутизмом, ранней детской шизофренией. Для синдрома Геллера характерно общее психическое опустошение.
Лечение Синдрома Геллера (симбиотического психоза, детской деменции, болезни Геллера - Цапперта):
Преимущественно симптоматическое. Включает три направления: лечение нарушений поведения и неврологических расстройств; мероприятия социальной и образовательной служб; помощь семье и семейную терапию.
К каким докторам следует обращаться если у Вас Синдром Геллера (симбиотический психоз, детская деменция, болезнь Геллера - Цапперта):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Геллера (симбиотического психоза, детской деменции, болезни Геллера - Цапперта), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Геллера-Цапперта болезнь
Геллера-Цапперта болезнь - прогрессирующее слабоумие неустановленной этиологии, развивающееся у детей в возрасте после 2-3 лет (Heller, Zappert, 1909). Ребёнок, согласно описаниям авторов, делается поначалу капризным, плаксивым, мрачным, возбудимым, затем его психическое развитие приостанавливается. В последующем быстрыми темпами развиваются слабоумие, оскудение речи, могут возникать беспричинные страхи, а также обманы восприятия. Спустя 0,5 -1 год от начала болезни речь полностью утрачивается, пациент становится мутичным, не понимает услышанной родной речи, не выполняет указаний, просьб. Одновременно с этим совершаются стереотипные кататоноподобные действия, наблюдаются насильственный смех и плач, иногда - эпилептиформные припадки. Определённой очаговой неврологической симптоматики, как правило, не выявляется. Лицо пациента неизменно сохраняет интеллектуальное выражение. Лечение симптоматическое, эффективных методов профилактики заболевания ныне не существует. Синоним: Dementia infantilis Helleri.
Энциклопедический словарь по психологии и педагогике . 2013 .
Полезное
Смотреть что такое "Геллера-Цапперта болезнь" в других словарях:
Геллера-Цапперта болезнь — (Heller F., Zappert J., 1909). Прогрессирующее слабоумие у детей раннего возраста. До 2 3 лет ребенок развивается правильно, затем, без видимой причины, становится возбудимым, сердится, беспричинно плачет, мрачен, угрюм. Постепенно развивается… … Толковый словарь психиатрических терминов
ГЕЛЛЕРА-ЦАППЕРТА БОЛЕЗНЬ — [Heller F., Zappert J., 1909]. Прогрессирующее слабоумие у детей раннего возраста. До 2 3 лет ребенок развивается правильно, затем, без видимой причины, становится возбудимым, сердится, беспричинно плачет, мрачен, угрюм. Постепенно развивается… … Толковый словарь психиатрических терминов
Синдром Геллера — - см. Геллера Цапперта болезнь … Энциклопедический словарь по психологии и педагогике
ОЛИГОФРЕНИЯ — - группа различных по этиологии и патогенезу заболеваний, основным проявлением которых служит врожденное или приобретенное в первые 3 года жизни слабоумие и затруднение социальной адаптации. Причиной олигофрении могут быть хромосомные аномалии,… … Энциклопедический словарь по психологии и педагогике
Геллера (-Цапперта) синдром
Прогрессирующая деменция у детей раннего возраста. Относится к расстройствам аутистического спектра (син.: дезинтеграционный психоз, первазивное дезинтегративное расстройство), к которым также принадлежат синдромы Аспергера, Каннера, Ретта. Характеризуется началом заболевания в 2-6-летнем возрасте с расстройств речи (нечёткость и смазанность, вплоть до лепетания), в дальнейшем речевые навыки утрачиваются. Наблюдается также утрата понимания речи. Музыкальный слух сохраняется дольше речи. Характерны беспокойство, тревога, приступы гнева и страха, стереотипность движений, изредка - кататония, насильственный смех и плач, судорожные припадки. В дальнейшем (приблизительно через год) развивается полная утрата навыков и знаний. Соматически дети остаются обычно здоровыми, при глубоком слабоумии лицо сохранят осмысленное выражение.
Этиология и патогенез, так же как и нозологическая самостоятельность синдрома, окончательно не уточнены. Поражение локализуется преимущественно в моторных центрах речи и «немых» моторных областях головного мозга (лобно-височные отделы). При ПЭГ и КТ выявляют гидроцефалию и корковую атрофию. Дифференциальный диагноз проводят с афазией и шизофренией у детей и эпилептической деменцией.
Теодор Геллер, австрийский психиатр, педагог и детский психолог, в 1908г. описал 6 случаев синдрома, названного им «dementia infantilis» (юношеской деменцией) (Heller T. Über Dementia infantilis: Verblödungsprozeβ im Kindesalter // Zeitschrift für die Erforschung und Behandlung des Jugendlichen Schwachsinns, 1908. - Bd.2. - S.17-28). Юлиус Цапперт, австрийский педиатр, в 1911г. уточнил клиническую картину синдрома.
Узники собственного тела: синдром «взаперти»

Почти в любом сериале, посвященном работе врачей, есть эпизоды, связанные с лечением больного с синдромом «запертого человека» (Locked-in-syndrome). Что это за заболевание, и почему оно привлекает столько внимания, больше, чем другие редкие болезни? MedAboutMe предлагает разбираться вместе.
Заперты в собственном теле: псевдокома
В отличие от истинной комы, «запертый» человек находится в полном сознании. Но во многих случаях не может никак сообщить об этом окружающим, так как утрачивает способность управлять собственным телом. Врачи выделяют три степени синдрома запертого человека (СЗЧ):
- Полный СЗЧ. В этом состоянии нарушены все двигательные волокна, идущие от головного мозга, а также расположенные в стволе головного мозга центры дыхания, глотания, речи и мимики. Помимо тетраплегии (паралича всех четырех конечностей), у больного отсутствует возможность двигать всеми остальными мышцами тела, включая даже мышцы, обеспечивающие моргание и движение глаз.
- Классический СЗЧ. Все, что описано выше, за исключением вертикального движения глаз и моргания. Это дает возможность для общения с помощью специального кода, простейшим является код «Да-Нет», в котором утвердительному ответу соответствует одинарное моргание, отрицательному — двойное.
- Неполный СЗЧ. Помимо движения глаз, могут сохраняться или быстро восстанавливаться некоторые другие движения. Обычно это движения головы или всех или некоторых пальцев на руках и ногах, или движения мимических мышц.
Во всех случаях электроэнцефалограмма показывает нормальную активность мозга, но гораздо более показательным методом диагностики является позитронно-эмиссионная томография (ПЭТ). Болевая чувствительность может полностью отсутствовать или частично сохраняться.
По имеющейся статистике, чаще всего СЗЧ диагностируется у людей 40-50 лет, половая принадлежность не играет роли. Чем моложе больной, тем больше у него шансов на восстановление, особенно при адекватной терапии.
У врачей нет достаточно полной информации относительно редкости синдрома «запертого человека», так как, по всей видимости, многие случаи остаются недиагностированными. До 2009 года в медицинской литературе было описано всего 33 случая болезни. А это означает, что «запертые» в своем теле пациенты не получали того лечения, которое помогло бы им восстановиться хотя бы частично. А это возможно благодаря современным возможностям медицины. Еще недавно 80% пациентов с синдромом «взаперти» умирали в течение первых 6 месяцев после начала заболевания. Сегодня пятилетняя выживаемость — более 60%. Причем сами больные отмечают удовлетворительное качество своей жизни, несмотря на все имеющиеся ограничения возможностей.
Причины развития синдрома
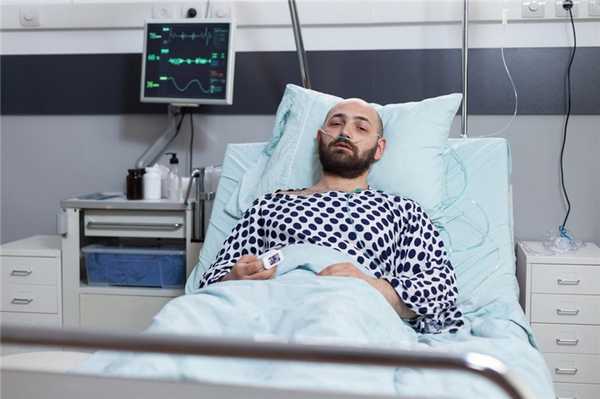
Чаще всего СЗЧ развивается вследствие инсульта с локализацией очага поражения в мосте головного мозга.
Мост представляет собой подковообразное скопление нервных волокон, соединяющее продолговатый мозг и мозжечок, среди функций которого координация движений и управление двигательными функциями тела.
Другими причинами могут стать инфекционные заболевания мозга, синдром Гийена-Барре, боковой амиотрофический склероз (БАС), рак мозга с расположением опухоли в области задней черепной ямки, а также серьезные черепно-мозговые травмы.
Клинический случай: Рикке Кьергаард
В 2013 году, во время рождественских праздников, живущая в Великобритании Рикке Кьергаард внезапно почувствовала себя плохо. Всего за четверть часа ее температура поднялась до 42°С, женщина начала бредить. На следующий день она уже не могла даже сесть без помощи окружающих, и вскоре впала в кому. В отделении больницы скорой помощи у Рикке была обнаружена полиорганная недостаточность, тромбоз множества сосудов, токсический шок, поставлен диагноз бактериальный менингит. Сердце женщины останавливалось на 40 секунд, но реанимационные мероприятия оказались успешными, и сердцебиение возобновилось, но сознание не вернулось. На тот момент врачи оценивали шанс Рикке выжить как очень низкий — не выше 5%.
Женщина пробыла в состоянии комы 10 дней.
Но потом сознание начало к ней возвращаться. И она услышала, как врачи говорят ее мужу, что ему стоит начать готовиться к похоронам, так как шансов на то, что мозг его жены не умер, близки к нулю.
Рикке повезло: ее семья верила в нее. В какой-то момент муж Питер обратил внимание на то, что на вопросы жена отвечает морганием. Это стало сигналом, что функции мозга сохранились, что Рикке не только слышит, но и может общаться.
Процесс восстановления был долгим и трудным, ведь женщине пришлось не только заново учиться дышать, глотать, жевать, говорить, ходить, но и пережить ампутацию почти всех пальцев на руках из-за развившейся гангрены, и частичную потерю зрения: один глаз перестал видеть. Через 4 месяца Рикке смогла произнести первое слово.
Восстановившись, насколько это было возможно, Рикке Кьергаард основала компанию, главная цель которой — помогать людям с тяжелыми хроническими заболеваниями. А еще она написала книгу о своем удивительном опыте узника собственного тела.
Не всем «узникам» везет так же, как Рикке. Медицинский персонал или родственники (если они есть) могут не заметить проявлений сознания, особенно в случае полного СЗЧ, когда больной не может даже морганием дать понять, что слышит и осознает окружающее. Еще сложнее диагностировать СЗЧ, если у больного есть нарушения слуха и зрения.
Бельгийские исследователи сообщают в статье, опубликованной в 2005 году о том, что иногда диагностика синдрома требовала от 4 до 6 лет — пока окружающие улавливали признаки того, что больной находится в сознании. В той же статье указывается, что как только пациент после диагностики СЗЧ начинает получать адекватную медицинскую помощь, ожидаемая продолжительность жизни увеличивается до нескольких десятилетий, хотя шансы на восстановление двигательных функций могут оставаться низкими.
Лечение синдрома «взаперти»

Специфического лечения не существует, как и лекарств. В каждом случае терапия подбирается индивидуально, и направлена на устранение причин, вызвавших паралич. Однако замечено, что если в остром периоде заболевания начата интенсивная, или даже агрессивная терапия, шансов на восстановление больше. Известны случаи даже полного восстановления, но они очень редки. В большинстве случаев можно рассчитывать только на частичную реабилитацию.
На начальном этапе важнее всего поддерживающая терапия и обучение самого больного и окружающих навыкам общения с использованием имеющихся возможностей.
Проблемы могут возникать на разных уровнях, начиная с дыхания и питания. Из-за нарушения дыхательных функций пациенты с СЗЧ часто находятся на аппарате искусственного дыхания, воздух поступает в легкие через трахеостому — специальную трубку, вводимую в дыхательные пути через отверстие в трахее. Питание через рот в этом случае представляет опасность, поэтому больного кормят через зонд. Переход к обычному питанию возможен только после восстановления дыхательной и глотательной функций, и только жидкой или пюреобразной пищей.
Из-за неподвижности больного высок риск развития пневмонии, тромбоза, инфекций мочевыводящих путей, образования пролежней и контрактур конечностей. Пациент нуждается в частой смене положения тела, в массаже, физиотерапевтическом лечении.
С помощью логопедов устанавливается система общения. Благодаря современным технологиям у больных с СЗЧ появилась возможность использовать для общения компьютер. Для этого используются специальные датчики, реагирующие на движения глаз, а также синтезаторы речи. Если сохранились или восстановились двигательные функции каких-либо мышц, то возможности еще более расширяются: такие больные могут использовать коляски с электроприводом, с управлением, приспособленным к имеющимся возможностям.
Прогноз излечения
Несмотря на то, что многие люди, столкнувшиеся с синдромом «взаперти», не выживают из-за несовершенной диагностики или развившихся осложнений, у остальных есть довольно много шансов не только выжить, но и прожить несколько десятков лет.
Вот что говорят проведенные исследования.
Анализ информации, полученной от самих больных и их родственников, показывает, что окружающие часто оценивают качество жизни больных существенно ниже, чем сами больные. То есть, несмотря на все ограничения, очень многие пациенты с СЗЧ оценивают качество своей жизни как удовлетворительное и даже хорошее. При этом оценка качества жизни прямо связана с уровнем оценки психосоциальной поддержки со стороны окружающих. Это дает основания для вывода о том, что тяжелая двигательная инвалидность не обязательно воспринимается человеком как невыносимая. Анкетирование больных СЗЧ показало, что подавляющее большинство ценят жизнь, хотят и намерены жить столько, сколько получится.
В интернете можно найти несколько книг, написанных людьми, пережившими «заключение» в собственном теле, или остающимися взаперти. Каждая из них — история успешной борьбы и победы, маячок для тех, кто ищет опору и мотивацию, знак, что не стоит сдаваться и «умирать раньше смерти».
Синдром Апера
Синдром Апера - генетическое заболевание, характеризующееся нарушениями процессов окостенения черепа и связанными с этим вторичными расстройствами, а также многочисленными пороками развития скелета и конечностей. Симптомами этого состояния являются карликовый рост, башенная форма черепа, расширенная переносица, незаращение твердого нёба, синдактилии на руках и ногах. Диагностика синдрома Апера производится по характерной клинической картине патологии, на основании рентгенологических данных и молекулярно-генетических исследований. Специфического лечения заболевания не существует, применяют поддерживающую терапию, проводят хирургические вмешательства паллиативного характера.
Общие сведения
Синдром Апера (акроцефалосиндактилия 1 типа) - генетическая патология, обусловленная нарушением образования некоторых видов соединительной ткани, главным образом костной. Впервые данное состояние было описано в 1906 году французским педиатром Э. Апером, дальнейшие исследования подтвердили генетическую природу этого заболевания. Этиология и молекулярно-генетические механизмы развития синдрома Апера были определены значительно позднее - лишь в 1995 году. Данная патология может наследоваться по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев ее причиной являются спонтанные мутации в половых клетках родителей (так называемые герминативные мутации).
Синдром Апера с одинаковой частотой поражает как мальчиков, так и девочек, его встречаемость составляет в среднем 1 случай на 160 000-200 000 новорожденных. Врачи-генетики в настоящее время относят синдром Апера к особой группе наследственных заболеваний - акроцефалосиндактилиям, характеризующиеся одновременным поражением костей черепа и конечностей. Особенностью этой патологии является важность ее как ранней диагностики, поскольку паллиативные мероприятия в раннем возрасте могут в значительной степени влиять на дальнейшее интеллектуальное развитие больного.
Причины синдрома Апера
Синдром Апера, согласно последним научным данным, обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Значительный размер (20 экзонов) и специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями. Помимо синдрома Апера дефекты гена FGFR2 приводят к развитию таких патологий, как синдром Бира-Стивенсона, синдром Пфайффера, синдром Сетре-Чотзена, краниофациально-скелетно-дерматологическая дисплазия и ряду других. Поэтому исследования данного гена довольно распространены в современной генетике.
Как показали исследования 1995-2000 годов, наиболее часто (в 96% случаев) к развитию синдрома Апера приводят мутации в области 7 экзона гена FGFR2. При этом на долю мутации S252W приходится порядка 74-76% от всех случаев заболевания, а примерно 21-23% вызываются дефектом P253R. Таким образом, причиной подавляющего большинства случаев синдрома Апера являются всего лишь два типа мутации, что упрощает молекулярно-генетическую диагностику этого состояния. Так как эти дефекты относятся к миссенс-мутациям, полученный в результате трансляции такого гена рецептор к фактору роста фибробластов имеет нарушенную структуру и неспособен выполнять свои функции. Это приводит к нарушению процессов окостенения черепа, в частности - к преждевременному зарастанию швов и остановке нормального роста черепной коробки. Дефект рецепторов при синдроме Апера также становится причиной пороков развития иных структур, где участвуют фибробласты (стенки сосудов крупного калибра, сердце, кости лицевого черепа, трахея). Наследуется это состояние по аутосомно-доминантному механизму, но чаще всего имеют место спонтанные мутации.
Кроме того, при синдроме Апера возникает аномальная экспрессия гена KGFR, тоже расположенного на 10 хромосоме. Он кодирует последовательность белка, являющегося рецептором к фактору роста кератоцитов. Никаких мутаций или других нарушений в структуре KGFR при синдроме Апера выявлено не было, лишь его чрезмерная активность, приводящая к увеличению количества кодируемых им рецепторов. Возможно, это явление объясняется сложными взаимоотношениями генов или же рецептор к фактору роста фибробластов 2 обладает супрессирующим действием на ген KGFR. Результатом аномальной экспрессии этого гена становятся фенотипические нарушения формирования конечностей - различные формы синдактилии, всегда встречающиеся при синдроме Апера, иногда полидактилия.
Симптомы синдрома Апера
Некоторые проявления синдрома Апера заметны с самого рождения - например, синдактилия, которая может быть полной или в виде перепонок. Как правило, срастаются 2, 3 и 4 пальцы на кистях, иногда аналогичный порок возникает и на пальцах ног. Среди неонатологов симптом иногда носит название «среднего пальца» - в тяжелых случаях эти три пальца прочно срастаются между собой и имеют один общий ноготь. Другим постоянным симптомом синдрома Апера, обнаруживающимся сразу после рождения или в первые месяцы жизни, является раннее развитие синостоза костей черепа. Чаще всего происходит срастание венечного или стреловидного шва, что по мере роста головного мозга приводит к деформации черепа по типу «башенной». Из-за черепного синостоза у больных синдромом Апера наблюдается хроническое повышение внутричерепного давления, становящееся причиной задержки умственного развития, головных болей, тошноты и рвоты.
Помимо деформации черепа о наличии синдрома Апера свидетельствует характерный внешний вид больных. У них обычно обнаруживается плоский или выпуклый лоб, гипертелоризм и экзофтальм, может развиваться косоглазие. Деформации затрагивают и кости лицевого черепа - переносица расширена, челюсти нередко недоразвиты, наблюдается нарушение прикуса. Из других симптомов синдрома Апера иногда регистрируются нарушения дыхания (из-за недоразвития верхней челюсти, сужения хоан или трахеи), незаращение твердого нёба, врожденные пороки сердца, аномалии развития позвонков, почек, прямой кишки.
У взрослых лиц, страдающих синдромом Апера, может возникать атрофия зрительных нервов вплоть до полной слепоты. Интеллектуальное развитие больных часто отстает от возрастной нормы, однако достоверно неизвестно, обусловлено это генетическими нарушениями или вторичными факторами (хронической внутричерепной гипертензией). Практически всегда при синдроме Апера наблюдается карликовый рост. При соответствующем паллиативном лечении и уходе больные могут доживать до преклонного возраста, но риск внезапной смерти из-за поражений дыхательной, нервной и сердечно-сосудистой систем у них намного выше, чем в популяции.
Диагностика
Диагностика синдрома Апера производится на основании осмотра и изучения настоящего статуса пациента, рентгенологических исследований, молекулярно-генетических анализов. При осмотре у больного выявляется синдактилия (у лиц старшего возраста могут обнаруживаться следы ее хирургической коррекции), деформация черепа - башенный череп или брахикефалия, характерный внешний вид лица.
С возрастом у больных синдромом Апера могут нарастать признаки нарушения дыхания, при ЭхоКГ нередко определяются пороки сердца и сосудов - стеноз легочного ствола или аорты, дефекты межжелудочковой перегородки. Иногда на этом фоне выявляются признаки сердечной недостаточности. Также возможно наличие иных пороков развития - аномалий позвонков, глухоты, слепоты (из-за катаракты, пигментного ретинита, атрофии зрительных нервов), патологий почек и поджелудочной железы. Из-за столь широкого спектра возможных нарушений больные синдромом Апера нуждаются в тщательном и всестороннем медицинском обследовании.
Рентгенологическими методиками уже у маленьких детей можно обнаружить синостоз костей черепа в области венечного или стреловидного шва. В дальнейшем при помощи рентгенографии можно определить характерную для синдрома Апера деформацию черепной коробки, пороки развития костей лицевого черепа, аномалии позвонков и другие нарушения.
Наиболее достоверным диагностическим методом при этом состоянии является молекулярно-генетический анализ. Как правило, для выявления синдрома Апера производят секвенирование 7 экзона гена FGFR2, иногда используют менее затратные техники, ориентированные только на поиск наиболее распространенных мутаций (S252W и P253R), приводящих к этому заболеванию. Подобные методики более дешевые и быстрые в выполнении, обладают точностью на уровне 95%, возможно их использование в качестве пренатальной диагностики этого состояния. Подобный анализ особенно актуален, если посредством профилактических УЗИ у плода выявляются нарушения, предположительно связанные с синдромом Апера - пороки развития черепа, сердца, верхних или нижних конечностей.
Лечение синдрома Апера
Специфического лечения синдрома Апера на сегодняшний день не существует, однако паллиативные и симптоматические мероприятия могут значительно облегчить состояние больного и улучшить качество его жизни. Особенно важно как можно раньше диагностировать это заболевание по той причине, что своевременная хирургическая коррекция черепного синостоза позволит избежать значительного роста внутричерепного давления. По многочисленным данным, после таких операций, произведенных в раннем детстве, признаки умственной неполноценности у больных синдромом Апера были выражены значительно слабее, иногда сохранялся нормальный интеллект. Поэтому борьба с внутричерепной гипертензией играет центральную роль в паллиативном лечении этого состояния. Если же у пациентов имеется умственная отсталость, то ее выраженность снижается путем психокоррекционной работы.
Другой часто выполняемой паллиативной хирургической операцией при синдроме Апера является вмешательство для разделения сросшихся пальцев на руках и ногах. Это относительно несложная процедура при перепончатом типе сращения, однако при более тяжелых формах порока операция значительно усложняется. При синдроме Апера также может потребоваться помощь хирургов в случае пороков сердца, сужения хоан или трахеи, нарушения формирования прямой кишки и других проявлений этого генетического заболевания. Больные нуждаются в регулярных медицинских обследованиях у специалистов различного профиля.
Прогноз и профилактика
Прогноз синдрома Апера неопределенный по причине очень широкого спектра проявлений и значительного диапазона их выраженности. На прогноз также оказывают влияние такие факторы, как своевременность диагностики заболевания, объем паллиативного и симптоматического лечения. При относительно легких случаях синдрома Апера или правильной терапии этого состояния больные могут доживать до преклонного возраста. При этом возможно снижение интеллекта и появляющиеся с возрастом нарушения все новых органов и систем, что негативно сказывается на качестве жизни пациентов. В тяжелых случаях наблюдается летальный исход в раннем детстве из-за врожденных пороков сердца или полиорганной недостаточности.
Профилактика синдрома Апера возможна только в качестве пренатальной диагностики, которая может производиться как ультразвуковыми методиками, так и путем молекулярно-генетического анализа. Обычно проявления патологии сначала обнаруживаются на профилактических УЗИ, а затем диагноз подтверждается врачом-генетиком. Если данное состояние удается выявить на ранних сроках беременности, то ставится вопрос о ее прерывании.
Читайте также: