Синдром Гертвига-Вейерса (Hertwig-Weyers) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 22.01.2026
Окуло-денто-дигитальная дисплазия - наследственное, преимущественно аутосомно-доминантное заболевание, вызываемое мутациями в генах ODDD, SDTY3, ODOD, картированы в локусе 6 q22- q24. Основными пороками глаз являются: микрофтальмия, микрокорнеа, помутнение роговицы, врожденная катаракта, гипоплазия переднего мезодермального листка радужки, эксцентричное расположение зрачка, офтальмогипертензия или глаукома, псевдогипертелоризм, эпикантус, птоз [3].
Общие проявления синдрома - микроцефалия с многочисленными пороками развития: широкая переносица, утолщенная нижняя челюсть, расщелины нёба и верхней губы, маленький нос с широким корнем, острым кончиком, гипоплазия крыльев и узкие носовые ходы, дисплазия зубной эмали и другие аномалии зубов, а также разнообразные скелетные аномалии: короткие пальцы на руках и ногах, синдактилия и камптодактилия IV-V пальцев кисти, гипоплазия или аплазия средних фаланг одного или нескольких пальцев, уплощение I-V пястных костей, врожденный вывих бедра, расширение метафизов длинных трубчатых костей. Психомоторное развитие чаще нормальное, но у некоторых больных возможна умеренная умственная отсталость. Иногда наблюдается микроцефалия, микрогнатия, расщелины губы и неба, глухота, врожденный вывих бедра.
Дифференциальную диагностику окуло-денто-дигитальной дисплазии необходимо проводить с зубо-лицевым синдромом, сочетанием аномалии радужки и зубов. Лечение симптоматическое. По показаниям проводят удаление катаракты, антиглаукомные и челюстно-лицевые операции. Прогноз для зрения неблагоприятный 3.
В настоящей работе представлены результаты наблюдения за семьей пациентов (мать и ее ребенок) с окуло-денто-дигитальным синдромом (Лохманна-Мейер-Швиккера-Грютериха-Вейерса) в течение 8 лет - с 2011 по 2019 годы.
Семья больных, мама и ребенок, наблюдаются у офтальмолога с 2011 года, когда пациентка, 24 года, в перинатальном центре РКБ на сроке 38 недель беременности была впервые им осмотрена. Беременная - инвалид детства 1 группы из-за врожденной аномалии конечностей в виде наличия 2 пальцев на руках и ногах - тетраэктродактилии и слепоты. Со слов пациентки, у неe с раннего детства отмечалось низкое зрение. Так, в год у нее была выявлена миопия высокой степени (-15,0 Д), нистагм, а в 4 года развилась отслойка сетчатки на обоих глазах, по поводу которой ее не оперировали. В 12 лет диагностирована вторичная глаукома (компенсирована на каплях) и атрофия зрительного нерва, а в 17 лет - осложненная катаракта обоих глаз.
Что касается наследственности, то она оказалась отягощена. В частности, у отца и тети (сестры отца) диагностирована тетраэктродактилия, катаракта, отслойка сетчатки обоих глаз, а у матери, со слов беременной, - микрофтальм. Муж пациентки является инвалидом по зрению после перенесенной травмы с последующим развитием симпатической офтальмии.
При осмотре выявлено: лицо ассиметричное, вытянуто книзу - большая нижняя челюсть, маленькая верхняя - микродонтия, тусклые серые зубы - гипоплазия эмали, широкая переносица. Волосы тонкие, сухие, редкие. Кожа сухая. По 2 пальца на руках и ногах по типу клешни краба.
Острота зрения обоих глаз - 0, внутриглазное давление пальпаторно в норме, постоянный ротационный нистагм. Глазные яблоки увеличены в размере, имеются облаковидные помутнения нижней половины роговицы. Передняя камера мелкая, радужная оболочка субатрофичная, зрачки диаметром 6 мм, не реагируют на свет, помутнения хрусталика в передних корковых слоях и под задней капсулой. В стекловидном теле фиксированные помутнения. Тотальная отслойка сетчатки.
В 2011 году на сроке 39-40 недель было проведено родоразрешение путем кесарева сечения. Родился ребенок: пол женский, вес 2 кг 760 гр, рост 54 см. Голова деформирована, конечности короткие, искривлены, недоразвитые, имеется только по 1 пальцу на руках и ногах.
При осмотре органа зрения новорожденной было выявлено: передний отрезок глаз не изменен, роговица, хрусталик, передние слои стекловидного тела прозрачные. Рефлекс с глазного дна серый. При ультразвуковом исследовании обнаружены преретинальный фиброз стекловидного тела, закрывающий область диска и макулы, плоская субтотальная тракционная отслойка сетчатки обоих глаз.
Ребенок с рождения и до настоящего времени наблюдается офтальмологом. В 2012 году ребенок с мамой были проконсультированы в Московском НИИ глазных болезней им. Гельмгольца. Диагноз: «врожденная патология - окуло-денто-дигитальная дисплазия, нистагм, косоглазие, плоская отслойка сетчатки, частичная атрофия зрительного нерва обоих глаз». Острота зрения соответствовала светоощущению с неправильной проекцией. В 2013 и 2014 годах году ребенку была проведена лазеркоагуляция сетчатки. В 2015 году при ультразвуковом исследовании обнаружена двусторонняя тотальная отслойка сетчатки, атрофия зрительного нерва. При дальнейшем обследовании светоощущение уже не определялось.
Дальнейшее наблюдение за пациенткой и ее ребенком констатировало стабилизацию состояния органа зрения. Острота зрения девочки - 0, внутриглазное давление нормальное, тотальная двусторонняя отслойка сетчатки, полная атрофия зрительного нерва. Лицо ребенка такое же, как у мамы: ассиметричное, с большой нижней челюстью и маленькой верхней, широкой переносицей и недоразвитыми зубами. Голова вытянутой формы, покрыта редкими тонкими волосами. На коротких конечностях по одному пальцу.
На примере приведенного клинического случая наблюдения за пациенткой и ее ребенком с окуло-денто-дигитальной дисплазией (синдромом Лохманн-Мейер-Швиккера-Грютериха-Вейерса) видно, что, учитывая наследственный характер заболевания, рекомендуется проводить тщательную пренатальную диагностику для выявления патологии эмбриона на минимальных сроках внутриутробного развития и рекомендовать прерывание беременности на ранних сроках по медицинским показаниям, поскольку патология со стороны органа зрения и конечностей необратима и не поддается лечению.
Мейер-Швиккера-Грютериха-Вейерса синдром
Окуло-денто-дигитальная дисплазия (синдром Мейер-Швиккера-Грютериха-Вейерса (G. Meyer-Schwickerth, Е Gruterich, H. Weyers, 1957; W. Lohmann, 1920)) - наследственное, преимущественно аутосомно-доминантное заболевание, вызываемое мутациями в генах ODDD, SDTY3, ODOD, картированы в локусе 6 q22- q24.
Клиническая симптоматика:
- характерны микроцефалия с многочисленными аномалиями дисгенеза (широкая переносица, иногда - утолщённая нижняя челюсть, расщелины неба и верхней губы, маленький нос с широким корнем, острым кончиком, гипоплазией крыльев и узкими носовыми ходами, узкие глазные щели, эпикантус, птоз, микрофтальм с микрокорнеа),
- генерализованная дисплазия зубной эмали и другие аномалии зубов, а также разнообразные скелетные аномалии (короткие пальцы на руках и ногах, синдактилия и камптодактилия IV-V, гипоплазия или аплазия средних фаланг одного или нескольких пальцев, уплощение I-V пястных костей, врожденный вывих бедра, расширение метафизов длинных трубчатых костей);
- патологические изменения глаз у этих больных включают
- различные аномалии радужки,
- персистирующую зрачковую мембрану
- аномалии гониодисгенеза (на этом фоне у них нередко развивается глаукома),
- часто - врождённую катаракту,
- гипоплазию зрительного нерва и другие патологические изменения, с которыми связано очень низкое зрение;
Тип наследования - аутосомно-доминантный.
Клинический случай
В мае 2011 года в роддоме офтальмологом была консультирована беременная, 24 лет, на сроке 38 недель, беременность вторая, является инвалидом 1 группы с детства из-за врожденной аномалии конечностей в виде наличия 2 пальцев на руках и ногах - тетраэктродактилии и слепоты.
Наследственность отягощена: у отца и тети (сестры отца) тетраэктродактилия, катаракта, отслойка сетчатки обоих глаз, у матери - односторонний микрофтальм. Муж - инвалид 1 группы по зрению (травма, симпатическая офтальмия), соматически здоров.
Из анамнеза было выявлено, что в 2008 году, во время первой беременности на малом сроке были выявлены множественные пороки развития плода - аномалии пальцев кистей и стоп по типу клешни краба (наследственный аутосомно-доминантный окуло-денто-дигитальный синдром), буфтальм, отслойка сетчатки. Было проведено прерывание беременности по медицинским показаниям. При настоящей беременности пациентка также была обследована на сроке 14 недель, также были выявлены множественные пороки развития плода. Но от прерывания беременности на этот раз категорически отказалась.
Объективный осмотр: лицо ассиметричное, вытянуто книзу - большая нижняя челюсть, маленькая верхняя - микродонтия, тусклые серые зубы - гипоплазия эмали, широкая переносица. Волосы тонкие, сухие, редкие. Кожа сухая. По 2 пальца на руках и ногах.
Status oculorum: Visus OU=0. OU - горизонтально-ротационный нистагм, «буфтальм», множественные склеральные стафиломы, вторичная компенсированная глаукома, тотальные помутнения роговицы. Рефлекса с глазного дна нет.
Далее произведено родоразрешение путем кесарева сечения. Родился доношенный ребенок: пол женский, вес 2 кг 760 гр, рост 54 см с врожденной патологией. У ребенка по 1 пальцу на руках и ногах, окуло-денто-дигитальная дисплазия, нистагм, косоглазие, преретинальный фиброз, плоская отслойка сетчатки, атрофия зрительного нерва обоих глаз. Отмечалась двигательная активность на световой раздражитель, взгляд не фиксирует. В дальнейшем наблюдались нерегулярно, зрение было - неуверенное светоощущение, проведена лазеркоагуляция сетчатки, были на осмотре пару раз, status idem.
Через 2 года - у мамы - полная слепота (зрение=0), компенсированная вторичная глаукома. У девочки зрение = светоощущение, фиброз стекловидного тела, отслойка сетчатки, ВГД норма.
Синдром Уивера у ребенка 16 лет (описание клинического случая)
В практической деятельности педиатр нередко наблюдает больных со значительными отклонениями в физическом развитии. В большинстве обращений эти изменения касаются низкорослости и довольно часто обусловлены наследственными факторами. Но особого внимания зас
Weaver syndrome in a 16-year-old child (clinical case description) / T. A. Bokova, D. A. Kartashova, Yu. Yu. Kotalevskaya
In their practice, pediatricians often observes patients with significant disorders in their physical development. In most cases, these changes are related to dwarfism and quite often conditioned by hereditary factors. But special attention should be paid to children with high anthropometric parameters. The article presents a case of own observation of extremely rare combination of tallness and congenital disorders in development - Weaver syndrome in a 16-year-old adolescent.
В 1974 г. американские генетики, возглавляемые известным ученым, профессором медицинской и молекулярной генетики Медицинской школы Университета Индианы D. Weaver, впервые опубликовали случай данной патологии у двух мальчиков [1]. До настоящего времени в мире описано не более 50 наблюдений этого синдрома. Из ныне живущих больных с синдромом Уивера известна 22-летняя турчанка Румейса Гелги (Rumeysa Gelgi), которая занесена в книгу рекордов Гиннеса как самая высокая женщина в мире. Ее рост составляет 2 метра 15 см. В РНПЦ «Мать и дитя» Минздрава Республики Беларусь в 2013 г. опубликовано 5 случаев наблюдения данного синдрома [2]. В отечественной литературе найдено описание клинического примера 5-летней девочки, наблюдавшейся в Московском НИИ педиатрии и детской хирургии Минздрава РФ в 2000 г. [3].
Тип наследования синдрома Уивера до сих пор остается неясным. Большинство описанных случаев были спорадическими. Проявления синдрома Уивера характеризуются вариабельной экспрессивностью и большей частотой среди лиц мужского пола.
Диагностика заболевания состоит из следующего симптомокомплекса: высокие показатели физического развития (SDS роста > 2), типичные краниофасциальные симптомы, низкий хриплый голос, изменения опорно-двигательного аппарата, поражение органов зрения, возможна задержка психомоторного и речевого развития, часто нормальный уровень соматомедина С и гормона роста, костный возраст соответствует паспортному или опережает его.
Генетическая основа синдрома Уивера длительное время оставалась неизвестной. В 2003 г. J. Douglas с соавт. описали мутации в гене NSD1, отвечающем за развитие синдрома Сотоса, у 3 пациентов из 7 с диагнозом «синдром Уивера» [4]. Однако в 2012 г. было проведено полное секвенирование экзома неродственным пациентам с синдромом Уивера и в результате выявлены мутации в гене EZH2, расположенном на 7-й хромосоме [5].
Дифференциальная диагностика проводится с другими заболеваниями, сопровождающимися макросомией и аномалиями развития: в первую очередь с синдромом Сотоса, синдромом Видемана-Беквита, синдромом МОМО, синдромом Симпсона-Голаби-Бемеля, полисомией Y, синдромом Вермера, синдромом Карнея, синдромом Клайнфельтера, синдромом Марфана, гомоцистинурией, синдромом Пайла, синдромом Маршалла-Смита и другими.
Клиническое наблюдение
Мальчик поступил в педиатрическую клинику МОНИКИ впервые в возрасте 15 лет с жалобами на высокий рост (за последний год прирост 15 см), увеличение размера ноги за год с 44 до 48, деформацию позвоночника и грудной клетки, ухудшение зрения.
Ребенок от второй беременности (первый ребенок здоров), протекавшей на фоне повторных угроз прерывания на ранних сроках. Роды срочные, самопроизвольные. Масса при рождении 4200 г, длина 57 см. С рождения физическое развитие гармоничное, высокое. Психомоторное развитие и половое развитие соответствуют возрасту. Наблюдается офтальмологом по поводу гиперметропии высокой степени обоих глаз, хирургом-ортопедом по поводу нарушения осанки, деформации грудной клетки, плоскостопия, челюстно-лицевая хирургия по поводу микрогнатии (получает лечение в виде дистракционного остеосинтеза нижней челюсти с двух сторон) и ортодонтом по поводу нарушения прикуса и роста зубов. Рост отца и матери ребенка 172 см. У родственников мужского пола по линии матери рост 180 см — 2 м.
С рождения отмечались большие прибавки роста. При обследовании в 2 года — физическое развитие на 4 года. Общеклинические анализы в норме. Гормон роста в пределах референсных значений. Костный возраст на 3-4 года. МРТ головного мозга — без патологии. Ребенок и в детском саду, и в школе был значительно выше своих сверстников.
С 2012 г. появились жалобы на головокружение, головную боль, периодический акроцианоз, потерю сознания при изменении положения тела. При мониторинге артериального давления выявлена склонность к гипотонии. Предъявляемые ребенком жалобы объяснялись отставанием развития сердечной мышцы и сосудистой системы по сравнению со скоростью роста костей, тогда как сама причина такого стремительного роста оставалась неясной.
В 2017 г. мальчик поступил в педиатрическое отделение МОНИКИ с направительным диагнозом: «Синдром высокорослости».
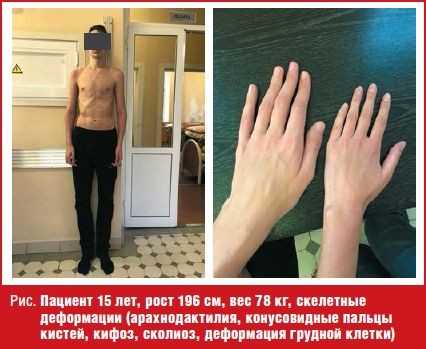
При поступлении рост мальчика рост 196 см, вес 78 кг. Обращали на себя внимание: грубый хриплый голос, лицевые дизморфии (антимонголоидный разрез глаз, удлиненные глазные щели, широкий и короткий нос, широкий длинный фильтр, ретромикрогнатия, большие диспластичные ушные раковины), скелетные деформации (арахнодактилия, конусовидные пальцы кистей, кифоз, сколиоз, деформация грудной клетки) (рис.). По лабораторным данным: общий анализ крови, мочи, кала, развернутый биохимический анализ крови, гормональный профиль (гормоны щитовидной железы, гипофиза, надпочечников, половые гормоны), исследование фосфорно-кальциевого обмена — показатели в пределах референсных значений. ЭКГ, Эхо-КГ, УЗИ брюшной полости, забрюшинного пространства и щитовидной железы — без особенностей. Уровень артериального давления в пределах возрастной нормы. МРТ головного мозга и гипоталамо-гипофизарной области прицельно — без патологии. Рентгенограмма кистей — костный возраст соответствует 15,5-16 годам. Осмотрен совместно с узкими специалистами (эндокринолог, генетик): SDS роста +3,4. Половое развитие соответствует возрасту. Высокорослость. Марфаноподобный синдром.
![Пациент 15 лет, рост 196 см, вес 78 кг, скелетные деформации]()
В результате комплексного обследования в клинике эндокринная причина избыточного роста ребенка была исключена, и тогда было принято решение провести углубленное генетическое обследование. Проведен поиск мутаций, ассоциированных с синдромом Марфана и другими наследственными заболеваниями со сходными фенотипическими проявлениями. В результате молекулярно-генетического анализа у мальчика выявлена мутация в 17-м экзоне гена EZH2 в гетерозиготном состоянии. Мутация в гене EZH2 является типичной для синдрома Уивера, однако выявленная у пациента мутация ранее не была описана.
Таким образом, учитывая фенотипические особенности пациента и наличие мутации в гене EZH2, был установлен диагноз «синдром Уивера».
Спустя год ребенок обследован в клинике повторно. Рост 2,01 м (годовая прибавка роста 5 см), вес 86 кг. Размер обуви остался без изменений. В ходе обследования общеклинические анализы, данные гормонального профиля, форфорно-кальциевого обмена, УЗИ брюшной полости, Эхо-КГ, мониторинг артериального давления, МРТ головного мозга, денситометрия в режиме позвоночник и все тело — без патологии. Костный возраст соответствует 16-16,5 годам.
Заключение
Несмотря на то, что все генетически детерминированные синдромы высокорослости встречаются крайне редко, характеризуются схожими проявлениями, не имеют специфического лечения, пациентам необходимо проводить комплексное генетическое обследование. В зависимости от генетического дефекта, различается спектр клинических проявлений различных синдромов, что позволяет прогнозировать в дальнейшем течение заболевания и осуществлять профилактические мероприятия по развитию некоторых осложнений. Так, например, учитывая, что мутации в гене EZH2 являются причиной синдрома Уивера, а ген EZH2 является белком гомеобокс группы, который участвует в поддержании гомеостаза, деления и дифференцировки клеток, соответственно, патологическая экспрессия этого гена у больных синдромом Уивера может быть причиной нейродегенеративных заболеваний и некоторых форм рака, например, нейробластомы. Кроме того, в гене EZH2 описаны мутации в соматических клетках при лейкемии, В-клеточных лимфомах и других онкологических заболеваниях крови, что обуславливает некоторую предрасположенность пациентов с синдромом Уивера к онкологическим заболеваниям 5. Эти сведения могут помочь лечащему врачу в определении тактики наблюдения пациента и осуществления профилактических мероприятий.
Литература
- Weaver D. D., Graham C. B., Thomas I. T., Smith D. W. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly // J. Pediatr. 1974; 84: 547-552.
- Ильина Е. Г., Ершова-Павлова А. А. Синдром Вивера в Беларуси // Здравоохранение (Минск), 2013; 3: 62-65.
- Казанцева Л. З., Семячкина А. Н., Новиков П. В., Новикова И. М., Добрынина Э. В. Синдром Вивера у детей // Российский вестник перинатологии и педиатрии. 2000; 2: 55-57.
- Douglas J. et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes // Am J Hum Genet. 2003, Jan; 72 (1): 132-143.
- Gibson, W. T., Hood R. L. et al. Mutations in EZH2 cause Weaver syndrome // Am. J. Hum. Genet. 2012, 90: 110-118.
- Качанов Д. Ю., Шаманская Т. В., Шевцов Д. В. и др. Генетическая предрасположенность к нейробластоме у детей: собственные данные и обзор литературы // Онкопедиатрия. 2016; 3 (4): 277-287.
- Chase A., Cross N. C. Aberrations of EZH2 in cancer // Clin Cancer Res. 2011, May 1; 17 (9): 2613-2618.
Т. А. Бокова 1 , доктор медицинских наук, профессор
Д. А. Карташова
Ю. Ю. Коталевская, кандидат медицинских наукГБУЗ МО МОНИКИ им. М. Ф. Владимирского, Москва
Синдром Уивера у ребенка 16 лет (описание клинического случая)/ Т. А. Бокова, Д. А. Карташова, Ю. Ю. Коталевская
Для цитирования: Лечащий врач № 1/2019; Номера страниц в выпуске: 12-13
Теги: синдром Уивера, физическое развитие, отклоненияСиндром Свайера ( Полная дисгенезия гонад , Чистая дисгенезия гонад )
Синдром Свайера - это нарушение формирования пола, характеризующееся кариотипом 46XY, врождённой дисгенезией гонад при первично сформированных прочих женских гениталиях - влагалище, матке, фаллопиевых трубах. К признакам патологии относятся первичная аменорея, маскулинное телосложение, половой инфантилизм. Диагноз выставляется на основании анамнестических данных, результатов общего и гинекологического осмотра, интроскопических методов исследования органов малого таза, гормонального и молекулярно-генетического анализа. Лечение состоит из двух этапов - удаления неразвитых половых желёз и длительного применения заместительной гормональной терапии.
МКБ-10
![Синдром Свайера]()
Общие сведения
Синдром Свайера (полную, или «чистую» дисгенезию гонад) можно кратко охарактеризовать как женский фенотип при мужском генотипе. Заболевание названо по имени британского эндокринолога Джералда Свайера, описавшего его в 1955 году как случай мужского псевдогермафродитизма. Полная форма дисгенезии является несиндромальной (не сопровождается экстрагенитальными пороками развития), исключает двойственность полового развития (наличие мужских первичных половых признаков наряду с женскими), психологическое развитие происходит по женскому типу. Врождённая патология встречается в одном случае на 20 000 индивидов с мужским кариотипом и регистрируется чаще иных форм XY-дисгенезии гонад.
Причины
Этиология синдрома Свайера изучена недостаточно. На сегодняшний день известно, что чаще всего возникновение патологии связано с отсутствием или мутацией гена SRY, расположенного на коротком плече хромосомы Y и большей частью отвечающего за контроль формирования тестикул. Судя по наблюдениям семейных случаев заболевания, возможна причастность пока неизвестных X-сцепленных или аутосомных генов.
Факторы риска тоже до конца не установлены. Кроме общих мутагенных воздействий (ионизирующего излучения и интоксикаций, вирусных инфекций, несбалансированного или пониженного питания) и уже упомянутого наследственного отягощения предполагается, что вероятность патологии может находиться в прямой зависимости от возраста отца. Чаще всего проследить связи между каким-либо воздействием на организм во время гестации и развитием синдрома Свайера не удаётся.
Патогенез
Формирование репродуктивных органов происходит из мюллерова протока у женщин и вольфова - у мужчин. У эмбриона с мужским генотипом синтез мужских стероидных гормонов клетками Лейдига в эмбриональных семенниках обусловлен воздействием материнского хорионического гонадотропина. Клетки Сертоли стимулируют дифференцировку клеток Лейдига и прочих, продуцируют антимюллеров гормон, способствующий атрофии мюллерова протока. Их нормальная деятельность обуславливает развитие индивида мужского пола - с адекватной дифференцировкой тестикул, атрофией мюллерова протока.
Выраженные сбои этого механизма приводят к формированию из бипотентных зачатков женских репродуктивных органов, развитие которых не требует столь сложной регуляции. Созревание мужского эмбриона контролируется геном SRY. При его отсутствии или мутации нарушается деятельность клеток Сертоли, дифференцировка гонад не происходит, что влечёт развитие синдрома Свайера - фенотипически женского организма без полноценных яичников, способных в дальнейшем стимулировать развитие вторичных половых признаков, но с бесполезными, склонными к малигнизации зачатками желёз.
Симптомы
В допубертатном периоде патология протекает без каких-либо субъективных проявлений. В период пубертата для синдрома Свайера характерно отсутствие признаков полового созревания. Может отмечаться лишь скудное оволосение в области лобка и подмышек, однако нередко отсутствует и оно. Менархе не наступает, молочные железы не развиваются или выражены очень слабо. Тип телосложения при овариальной дисгенезии мужской - с широкими плечами, объёмной грудной клеткой, узким тазом.
У женщин с синдромом Свайера чаще наблюдается нормальный или выше среднего рост, развитая мускулатура, «тяжёлая» нижняя челюсть. Иногда возникает незначительная гипертрофия клитора, хотя обычно наружные гениталии несколько недоразвиты. Больные жалуются на первичное бесплодие, ощущение дискомфорта или боли ввиду недостаточного развития вагины при половом акте или гинекологическом осмотре.
Осложнения
Частым (у 20-60% пациенток) осложнением синдрома Свайера является развитие опухолей, по большей части исходящих из рудиментарного полового тяжа, которым, по сути, представлены дисгенетичные гонады - дисгермином, андробластом. Эти неоплазии нередко имеют злокачественный характер, возникают с обеих сторон (синхронно или метахронно), регистрируются в раннем репродуктивном, подростковом и детском возрасте.
К последствиям нелеченного синдрома Свайера также можно отнести раннее появление патологий, обусловленных дефицитом эстрогена у женщин - остеопороза, сердечно-сосудистых заболеваний (артериальной гипертензии, ишемической болезни сердца). Кроме того, для многих больных диагноз становится источником тяжёлого психологического страдания по поводу «утраты женственности», а у замужних женщин - и боязни распада семьи.
Диагностика
Диагностика синдрома Свайера осуществляется гинекологом при участии медицинского генетика. Чаще всего диагноз устанавливается в 14-15 лет при обращении к врачу по поводу отсутствия полового развития, иногда - позднее, по причине бесплодия. В некоторых случаях первым признаком заболевания и основанием для углублённого исследования является опухолевое образование рудиментарных половых желёз, случайно обнаруженное врачом или самой больной. Диагностические мероприятия включают:
- Клинический осмотр. В ходе общего осмотра выявляется отсутствие основных вторичных женских половых признаков на фоне нормального или ускоренного роста. При гинекологическом осмотре обнаруживаются нормально сформированные, но недоразвитые женские наружные половые органы. Косвенным подтверждением диагноза является наличие кровных родственниц с полной или частичной дисгенезией гонад.
- Лучевые методы исследования. Наиболее доступным и информативным методом диагностики является гинекологическое УЗИ. Ультрасонография позволяет обнаружить такие характерные для патологии объективные признаки, как гипоплазию матки, извитые, недоразвитые маточные трубы, тяжевидные «яичники» (иногда - с новообразованиями). В сомнительных случаях назначается МРТ.
- Гормональный анализ. Характерным признаком синдрома Свайера считается значительное повышение гонадотропных (фолликулостимулирующего, лютеинизирующего) гормонов в сыворотке крови, снижение уровня эстрадиола. Гестагеновая функциональная проба отрицательная, циклическая (эстроген-гестагенная) гормональная проба положительная.
- Генетическое тестирование. Применяется с целью верификации диагноза. Цитогенетическое исследование выявляет мужской кариотип (46, XY), а молекулярно-генетическое - отсутствие или повреждение гена SRY. При невозможности генетического обследования назначается гистологическое исследование биоптата яичников. Диагноз подтверждает соединительнотканная структура с отсутствием фолликулов.
Синдром Свайера дифференцируют с другими формами дисгенезии гонад - синдромом Шерешевского-Тёрнера, мозаицизмом 45,X/46,XY, синдромом Морриса (синдромом тестикулярной феминизации), а также с задержкой полового развития гипоталамо-гипофизарного, конституционального, идиопатического генеза. При подозрении на опухоль необходима консультация онкогинеколога, а для выявления центральных форм задержки полового созревания - эндокринолога.
Лечение синдрома Свайера
Консервативная терапия
Лечение заболевания направлено на предупреждение осложнений, нормализацию психоэмоционального статуса пациентки, по возможности - на реализацию репродуктивной функции. После установления диагноза осуществляется хирургическая операция (удаление рудиментарных гонад), затем назначается гормональная терапия. Раннее (с подросткового периода) начало лечения синдрома Свайера повышает вероятность вынашивания ребёнка.
- Заместительная гормональная терапия. Проводится чередованием эстрогенных и прогестиновых препаратов в течение менструальных циклов. Назначается с периода полового созревания до возраста климактерия. Лечение нормализует течение метаболических процессов, способствует развитию матки, женских половых вторичных признаков, гравидарной подготовке эндометрия.
- Коррекция нервно-психических нарушений. Психотерапевтическое воздействие на личность позволяет перестроить отношение больной к себе и ближайшему окружению, преобразовать собственные психологические установки, снять психоэмоциональное напряжение. Иногда лечение дополняется фармакотерапией (антидепрессантами, транквилизаторами).
При достаточном развитии матки возможно применение программы ЭКО с имплантацией донорских ооцитов, оплодотворённых спермой супруга пациентки. На протяжении вынашивания плода женщине назначается сначала поддерживающая, затем сохраняющая гормонотерапия. Родоразрешение осуществляется оперативно. В современных репродуктологии и акушерстве уже накоплен успешный опыт ведения беременности у больных с дисгенезией гонад (в том числе синдромом Свайера).
Хирургическое лечение
Оперативное вмешательство в объёме удаления рудиментарных гонад и маточных труб выполняется непосредственно после диагностики этой формы врождённой овариальной агенезии. Хирургическое лечение проводится с целью предотвращения новообразований, источником которых являются клетки дисгенетичных половых желёз. Консервативное лечение не рекомендуется начинать до хирургической операции, поскольку гормональная терапия повышает риск развития онкологических осложнений.
Прогноз и профилактика
При своевременно начатом рациональном лечении синдрома Свайера прогноз относительно продолжительности и качества жизни благоприятный. Некоторые пациентки могут реализовать детородную функцию с помощью вспомогательных репродуктивных технологий, описаны случаи повторных успешных беременностей. Первичная профилактика рождения девочек с XY-дисгенезией включает здоровый образ жизни во время беременности, исключение профессиональных и бытовых вредностей. Для раннего выявления и лечения заболевания следует подвергать генетическому обследованию монозиготных близнецов больных.
1. Аномалии половых хромосом при нарушениях формирования пола и репродукции человека. Автореферат диссертации/ Черных В.Б. - 2015.
3. Гинекология: национальное руководство/ под ред. Кулакова В.И., Савельевой Г.М., Манухиной И.Б.. - 2009.
Синдром Туретта - симптомы и лечение
Что такое синдром Туретта? Причины возникновения, диагностику и методы лечения разберем в статье доктора Диордиева Максима Борисовича, психиатра со стажем в 8 лет.
Над статьей доктора Диордиева Максима Борисовича работали литературный редактор Вера Васина , научный редактор Владимир Вожжов и шеф-редактор Маргарита Тихонова
![Диордиев Максим Борисович, психиатр, нарколог, психотерапевт - Екатеринбург]()
Определение болезни. Причины заболевания
Синдром Туретта (Tourette's syndrome) — это заболевание нервной системы, при котором возникают множественные двигательные и вокальные тики. Для постановки диагноза они должны присутствовать дольше года.
![Тики при синдроме Туретта]()
Впервые заболевание, похожее на синдром Туретта, было описано в 1486 году в книге «Молот ведьм». Там упоминался священник с моторными и вокальными тиками, считавшийся одержимым. В конце XIX века симптомы заболевания на примере нескольких пациентов описал вместе с коллегами французский невролог Жорж Жиль де ла Туретт, в честь которого и назван синдром [1] .
Обычно синдром Туретта проявляется уже в детстве, но часто заболевание выявляют поздно или не диагностируют вовсе, так как родители не обращают на тики должного внимания. Из-за этого маленькие пациенты не получают своевременной помощи и могут страдать не только от самих тиков, но и от психологических проблем, связанных с заболеванием. В Европе от возникновения первых симптомов синдрома Туретта до постановки диагноза проходит в среднем более 5 лет [2] .
Распространённость синдрома Туретта
Синдром Туретта очень распространён — он встречается примерно у 10 из 1000 детей. В России его диагностируют у 8 человек на 10 000 населения, им могут страдать до 5 % школьников [3] . Мужчины болеют чаще, чем женщины: соотношение между ними составляет примерно 3 к 1.
Причины синдрома Туретта
Синдром Туретта — это генетическое расстройство, которое передаётся от родителей. Однако точный механизм наследования и ген, ответственный за болезнь, не известны. Риск передачи заболевания ребёнку составляет около 50 %. В прошлом, в начале XX века, тики считались следствием психотравм, но современная медицина это отвергает, так как такое предположение не удалось доказать [4] . Психосоциальные факторы и аутоиммунные заболевания не являются причиной синдрома Туретта, но могут влиять на тяжесть течения болезни.
Существует теория, что недостаток магния в организме и связанные с ним нарушения обмена веществ могут влиять на развитие синдрома Туретта. Косвенным доказательством этого служит то, что препараты с некоторыми соединениями магния могут улучшать состояние больных. Однако большие исследования на эту тему не проводились [5] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Туретта
Синдром Туретта проявляется тиками — быстрыми, внезапными, повторяющимися навязчивыми движениями или произнесением звуков. Чаще всего они возникают у мальчиков в возрасте от 4 до 11 лет. Тяжесть тиков доходит до пика примерно в 10-12 лет и ослабляется в подростковом возрасте. Большинство тиков исчезают спонтанно, но примерно у 1 % детей они сохраняются во взрослой жизни [7] .
Выделяют две основные группы тиков:
- Моторные тики — это непроизвольные движения частей тела. Самый распространённый из них — усиленное моргание. Также могут возникать подпрыгивания, постукивания по себе, развороты и повороты тела, гримасы, плевки, нецензурная жестикуляция и повторения чужих движений (копропраксия и эхопраксия).
- Вокальные, или звуковые, тики — это навязчивое произношение звуков, реже слов. Может проявляться кашлем, покашливанием, кряхтением. Иногда таких детей ошибочно лечат от бронхитов, трахеитов и бронхиальной астмы. Синдром Туретта часто ассоциируется с копролалией — внезапным высказыванием нецензурных фраз или слов, которое зачастую сопровождается копропраксией. Однако копролалия возникает только у 10 % пациентов [6] . Помимо копролалии, они могут повторять чужие слова, собственное слово или фразу (эхолалия и палилалия).
При синдроме Туретта моторные тики обязательно сочетаются с вокальными. Если присутствуют моторные тики, но нет вокальных, то стоит заподозрить другие заболевания: органическое поражение головного мозга, эпилепсию, синдром дефицита внимания (СДВГ), обсессивно-компульсивное расстройство (ОКР).
У многих детей и подростков с синдромом Туретта также отмечается СДВГ, ОКР, повышенная агрессивность, тревожность и склонность к депрессиям.
При синдроме Туретта интеллектуальные способности не нарушаются. Дети с этим заболеванием могут сильно расстраиваться от подшучиваний своих сверстников. При эмоциональном напряжении, вызванном пристальным вниманием или насмешками окружающих, тики могут усиливаться.
Форма тиков при синдроме Туретта может меняться в течение суток или недели, например от лёгких единичных моторных тиков утром или в начале недели до сложных и множественных по вечерам или под конец учебной недели. Видимо, их выраженность зависит в том числе от психоэмоциональных нагрузок.
Иногда дети пытаются сдерживать тики, но такой контроль возможен лишь в некоторой степени. Когда ребёнок старается подавить тики, симптомы могут усилиться. Попытка сдержать тик вызывает выраженный дискомфорт, из-за чего возрастает тревога — тикозные движения, наоборот, немного успокаивают. Из-за стресса, тревожных состояний и усталости тики могут учащаться и усиливаться.
Патогенез синдрома Туретта
Патогенез синдрома Туретта до конца не изучен. Известно лишь, что расстройство вызвано генетическими причинами. Скорее всего, при определённых генетических факторах нарушается работа нейромедиаторных систем в подкорковых образованиях и лобной коре.
Помимо генетических факторов, в патогенезе может участвовать и органическое повреждение головного мозга, например при патологии беременности и родов, черепно-мозговых травмах или нейроинфекциях.
Основная роль в патогенезе заболевания, вероятно, принадлежит дисфункции лобных долей. Считается, что большую роль в развитии синдрома Туретта играет правая лобно-височная область, сенсомоторные отделы орбитофронтальной коры, моторная область, базальные ганглии и поясная извилина. Также важное значение имеют нарушения в кортико-стрио-таламо-кортикальном контуре — нейронных цепях, связывающих кору, базальные ганглии и таламус.
![Кортико-стрио-таламо-кортикальный контур]()
Нарушения в работе этих структур также характерны для детей с ОКР и СДВГ — эти заболевания часто сопутствуют синдрому Туретта. Даже известны генетически связанные с синдромом Туретта формы ОКР (преимущественно ОКР с навязчивыми действиями — F42.1).
Предположительно, при синдроме Туретта нарушается работа дофаминергической системы. Изменения, вероятно, затрагивают серотонин-, норадреналин-, глутамат-, холин-, ГАМКергическую и опиоидную системы. Косвенно на связь синдрома Туретта с дофамином указывает то, что тики уменьшаются при лечении препаратами, которые воздействуют на передачу нервного импульса, вызванную дофамином ( например, путём блокады постсинаптических D2-рецепторов) [2] .
Классификация и стадии развития синдрома Туретта
Синдром Туретта — это разновидность хронических тиковых (или тикозных) нарушений. В Международной классификации болезней (МКБ-10) заболевание кодируется как F95.2 Комбинированные голосовые и множественные двигательные тики.
В следующей Международной классификации болезней (МКБ-11) тики и синдром Туретта из психических расстройств перенесены в неврологические [1] .
Согласно классификации Американской психиатрической ассоциации (DSM-IV), тики подразделяются на следующие группы:
- по виду — двигательные или голосовые;
- по продолжительности — преходящие или хронические.
Преходящее тиковое расстройство — это множественные двигательные, голосовые или тики обоих видов, которые длятся от 1 до 12 месяцев. Хронические тиковые расстройства присутствуют больше года. Они могут быть одиночными или множественными, двигательными или голосовыми, но не оба вида сразу. Синдром Туретта относится к хроническому тиковому расстройству. Для постановки диагноза необходимо, чтобы множественные двигательные тики и хотя бы один голосовой тик наблюдались более года.
Стадии синдрома Туретта не выделяют. Но обычно расстройство начинается с преходящих двигательных тиков, как правило подёргивания лица, которые длятся до года. Часто это гримасничание, затем покашливание и шипение. Постепенно тики распространяются на руки, ноги и мышцы шеи. Затем, обычно через год, присоединяются вокальные тики, которые осложняют картину болезни.
Осложнения синдрома Туретта
Синдром Туретта может сопровождаться депрессией, тревожным расстройством, ОКР и СДВГ, что осложняет прогноз. Депрессия возникает из-за того, что детей с этим синдромом часто обижают и унижают. В результате у них формируется чувство одиночества и может развиться аутоагрессия, вплоть до попытки суицида. Поэтому важно не оставлять ребёнка один на один с этим расстройством: ему особенно необходима поддержка родителей и друзей.
При симптоме копролалии дети выкрикивают нецензурную брань, из-за чего могут подвергаться агрессии со стороны окружающих. Поэтому для таких пациентов очень важно организовать правильную социальную среду [7] .
Диагностика синдрома Туретта
Синдром Туретта диагностирует врач-психиатр или невролог, основываясь на наблюдении и сборе сведений об истории болезни, условиях жизни и перенесённых заболеваниях. Сейчас разрабатываются генетические карты, которые позволят с самого рождения определять совокупность генов, характерных для синдрома Туретта, но пока этот метод недоступен.
Чтобы установить диагноз «синдром Туретта», состояние должно соответствовать следующим критериям:
- присутствуют множественные двигательные тики и как минимум один голосовой тик;
- тики возникают много раз в день, почти ежедневно;
- расстройство длится более года, но необязательно непрерывно, ремиссии продолжаются меньше двух месяцев;
- симптомы появились в возрасте до 18 лет [8] .
Очень важно отличать тики при синдроме Туретта от вторичных тиков, которые появились на фоне инфекций или черепно-мозговых травм при беременности, родах или в раннем детстве. Их различают только на основе анамнеза и физикального обследования. Другие методы, например магнитно-резонансная томография и анализы крови, для диагностики синдрома Туретта не используются.
Лечение синдрома Туретта
Перед врачом всегда стоит выбор — назначать ли препараты при синдроме Туретта. При этом важно ориентироваться на состояние пациента, так как более чем в половине случаев симптомы исчезают без приёма медикаментов.
Психологическая помощь
Если тики не мешают человеку общаться, учиться и работать, то, скорее всего, принимать препараты не нужно. В такой ситуации будет полезно психологическое консультирование ребёнка и родителей. Важно рассказать родителям, что от ребёнка ни в коем случае нельзя требовать, чтобы он перестал кашлять и гримасничать, — это только усилит эмоциональное напряжение и, соответственно, тики.
В зарубежных странах, особенно в США, хорошо зарекомендовал себя метод под названием «Habit reversal training», т. е. тренировка отмены привычки [10] . Пациента учат отслеживать ощущение, предшествующее тикам, и пытаться заменить их на более приемлемые действия. Метод не избавляет от тиков, но заметно уменьшает их проявления.
Также для коррекции поведения используется когнитивно-поведенческая психотерапия [12] .
Медикаментозное лечение
Для лечения синдрома Туретта могут применяться антипсихотики (нейролептики) в невысоких дозировках, которые воздействуют на дофаминэргическую активность. Наиболее эффективны Арипипразол, Рисперидон и Галоперидол. Из них лучше всего переносится Арипипразол. Однако в российских инструкциях по лечению синдрома Туретта это лекарство не упоминается, хотя в США оно одобрено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) и активно используется в терапии. В России назначают Галоперидол и Рисперидон, а также ряд ноотропов (чаще всего гопантеновую и аминофенилмасляную кислоту) без достаточной доказательной базы.
Есть данные об эффективности Клонидина, но этот препарат опасен при передозировке. Среди возможных побочных эффектов — коллаптоидные состояния, т. е. резкое падение артериального давления, которое может привести к развитию обморока.
Существуют не подтверждённые данные об эффективности инъекции ботулотоксина в мышцы лица.
При возникновении сопутствующих заболеваний, таких как тревожно-депрессивное расстройство и ОКР, применяются селективные ингибиторы обратного захвата серотонина: Сертралин, Пароксетин, Эсциталопрам. При выраженной агрессии, направленной на себя или окружающих, назначаются нормотимики (препараты лития и другие) [12] .
Транскраниальная магнитная стимуляция
В настоящее время изучается влияние транскраниальной магнитной стимуляции на синдром Туретта как у детей, так и у взрослых, но пока недостаточно данных об эффективности этого метода [11] .
Нейрохирургическое лечение
При тяжёлом течении заболевания и выраженной устойчивости к медикаментам может применяться нейрохирургический подход — глубокая стимуляция подкорковых структур головного мозга (бледного шара и таламуса). Её используют для взрослых пациентов.
Прогноз. Профилактика
Прогноз благоприятнее и заболевание чаще заканчивается ремиссией или выздоровлением, если тики появились в возрасте до 7 лет.
Синдром Туретта часто вызывает у людей страх и ассоциации, что эта особенность мешает нормально общаться с другими. Но если грамотно подойти к терапии и исключить провоцирующие факторы, расстройство может протекать вполне благоприятно. Примером служит певица Билли Айлиш. Из-за тяжести заболевания она не ходила в школу и обучалась дома. Это не помешало певице в 2019 году записать сингл, завоевавший первые места в мировых хит-парадах, а в 2021 году войти в список 100 наиболее влиятельных людей года по версии журнала Time. Также синдромом Туретта, предположительно, страдал Вольфганг Моцарт [13] .
Синдром Туретта — это генетическое заболевание, поэтому предупредить его развитие нельзя. Однако если своевременно обратиться к врачу, можно снизить тяжесть болезни и предотвратить развитие депрессии, аутоагрессии и обсессивно-компульсивного расстройства.
При сильных тиках, не поддающихся лечению и мешающих общаться, учиться или работать, пациент может пройти медико-социальную экспертизу: специалисты оценят тяжесть состояния и, при необходимости, определят группу инвалидности.
Читайте также:
- Неиммунологические пути активации комплемента. Образование гранулем при аллергическом альвеолите
- Частота увеита у детей (эпидемиология)
- УЗИ, МРТ при стенозе сильвиева водопровода у плода
- Оценка почечной функции. Проба с фенолсульфофталеином.
- Задачи цитологического исследования. Установление природы частиц