Синдром Гоше (Gaucher) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Гоше болезнь (Ph. Ch. E. Gaucher, французский дерматолог, 1854— 1918) — наследственная болезнь, характеризующаяся накоплением глюкоцереброзидов в клетках системы фагоцитирующих мононуклеаров; относится к болезням накопления липидов — сфинголипидозам (см.).
Заболевание впервые описано Гоше в 1882 г. Гоше болезнь встречается редко.
Содержание
Этиология и патогенез
Гоше болезнь в большинстве случаев наследуется по аутосомно-рецессивному типу, поэтому чаще наблюдается у родных братьев, сестер. Описаны случаи Гоше болезни и у представителей разных поколений (дядей, тетей и племянников), когда наследование, по-видимому, идет по аутосомно-доминантному типу с неполной проявляемостью мутантного гена.
Развитие Гоше болезни обусловлено наследственным дефицитом гидролитического фермента глюкоцереброзидазы — бета-глюкозидазы (см. Глюкозидазы). Одним из источников накопления глюкоцереброзидов служат эритроциты. При разрушении эритроцитов в обычных местах фагоцитирования (селезенка, легкие) и расщеплении их липидной оболочки происходит освобождение глюкоцереброзидов. Еще более важным источником накопления глюкоцереброзидов служат распадающиеся лейкоциты. Дефицит фермента ведет к накоплению глюкоцереброзидов в клетках системы фагоцитирующих мононуклеаров и образованию т. о. клеток Гоше.
Патологическая анатомия
В разных органах обнаруживают мелкие или обширные очаги скопления клеток Гоше. Больше всего как при острой, так и при хронической форме болезни поражается селезенка. Она увеличена в размерах, нередко имеет бугристую поверхность. Ткань серо-красного, кирпичного или шоколадного цвета, на разрезе пестрая, с наличием ангиокавернозных очагов, инфарктов, рубцов. Микроскопически скопления клеток Гоше находят в красной пульпе, трабекулах, фолликулах; наблюдается резкое расширение синусов (но клеток Гоше здесь, как правило, нет). Клетки Гоше в селезенке содержат желто-коричневый пигмент. Печень также увеличена. Клеток Гоше здесь меньше, чем в селезенке; отдельные элементы представляют собой переходные формы от звездчатых эндотелиоцитов (купферовских клеток) к клеткам Гоше. Клетки Гоше располагаются диффузно в печеночных дольках, нарушая правильность их структуры, в стенках капилляров, вокруг синусов. В костном мозге очаговые скопления клеток Гоше сочетаются с рассасыванием костных балок, разрастанием соединительной ткани. В надпочечниках клетки Гоше находят преимущественно в ретикулярной зоне. В легких они инфильтрируют интерстициальную ткань альвеолярных перегородок, могут содержаться в просветах альвеол. При острой форме Г. б. эти клетки обнаруживают также в глиальных элементах головного мозга, тимусе, в клубочках почек. В головном мозге выявляют дистрофические изменения ганглиозных клеток, увеличение глиальных элементов в подкорковых узлах, явление нейронофагии (см.) и нарушение миелинизации. В белом веществе головного мозга иногда уменьшены отдельные фракции фосфолипидов, холестерина и цереброзидов, что может быть связано с демиелинизацией.
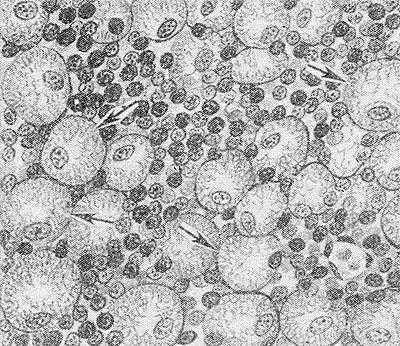
Клетки Гоше (рис. 1) округлой формы, крупные (диам. 40—80 мкм) с небольшим ядром, часто располагающимся эксцентрично, и широкой зоной фибриллярной, исчерченной светло-серой цитоплазмы. Для клеток Гоше характерна интенсивно-голубая цитоплазма при окраске по методу Маллори (см. Маллори методы), положительная ШИК-реакция (см.), высокая активность кислой фосфатазы и неспецифической эстеразы. В цитоплазме нередко содержится гемосидерин. Встречаются многоядерные клетки. При электронной микроскопии в клетках Гоше и в клетках, еще не приобретших морфол. черт клеток Гоше, но уже накапливающих глюкоцереброзиды, находят отсутствующие в норме липидные цитосомы — образования в виде скопления трубочек, содержащих глюкоцереброзиды, диам. 25— 40 нм, отграниченные мембраной. При использовании метода негативного контрастирования установлено, что эти трубочки имеют спиралевидную структуру. Это отличает клетки Гоше от сходных с ними клеток, выявляемых при других заболеваниях, в частности при хроническом миелолейкозе (см. Лейкозы). При электронной микроскопии в клетках Гоше нередко обнаруживают и остатки эритроцитов.
Клиническая картина
Различают две формы заболевания: острую, или злокачественную, и хроническую.
Злокачественная форма Гоше болезни проявляется в первые месяцы жизни ребенка. Характерно нарастание тяжести симптомов. Отмечают прогрессирующее увеличение селезенки и печени, отставание в физ. и психическом развитии, поражение ц. н. с.; развивается гипохромная анемия, лейкопения, тромбоцитопения; последняя, по-видимому, обусловливает появление кожных геморрагий, носовых кровотечений.
Поражение ц. н. с. проявляется увеличением размеров головы, умеренно выраженными признаками внутричерепной гипертензии (см. Гипертензивный синдром); отмечается также сходящееся косоглазие, гипертонус мышц шеи и гипотония (реже повышение тонуса) мышц конечностей, пирамидные симптомы. Характерными признаками в дальнейшем являются деменция (см. Слабоумие), отсутствие фиксации взгляда, ригидность мышц, сменяющаяся опистотонусом (см.).
Некоторые авторы считают характерными признаками острой формы Г. б. сочетание косоглазия, дистонии мышц конечностей и гиперэкстензии головы, деменции. Глазное дно обычно остается без изменений; у некоторых больных выявляется пигментный ретинит. Описанная неврологическая симптоматика может быть ведущей в клин, картине заболевания. Иногда ведущим в клин, картине острой формы Г. б. становится легочное поражение, обусловленное скоплением клеток Гоше в стенках альвеол. В этих случаях отмечается коклюшеподобный кашель, в мокроте можно найти клетки Гоше.
Хроническая форма протекает значительно доброкачественнее. В большинстве случаев она проявляется в юношеском возрасте или у взрослых. Физ. и психическое развитие больных, как правило, не страдает. Размеры селезенки и печени увеличиваются постепенно. Обычно в процесс вовлекаются и лимфатические узлы, преимущественно висцеральные. Изменения картины крови при хронической форме Гоше болезни те же, что и при злокачественной форме: анемия, лейкопения, тромбоцитопения; однако выражены они значительно слабее. Геморрагический синдром длительное время проявляется склонностью к подкожным кровоизлияниям и непродолжительными носовыми кровотечениями. Иногда отмечается субфебрильная температура. Характерно появление желто-коричневой пигментации на открытых частях кожи, слизистых оболочках и на склерах. Часто возникают боли в костях, вызывающие затруднения при ходьбе.
В ряде случаев костные поражения могут быть наиболее ранними симптомами и преобладать во всей клин, картине болезни. Иногда костные изменения осложняются патологическими переломами. У некоторых больных отмечается специфическое поражение легких и жел.-киш. тракта.
При хрон, форме Г. б. неврол, нарушения обнаруживают значительно реже, чем при острой, и выражены они весьма незначительно (пирамидные симптомы, интенционный тремор, дисметрия, вегетативные нарушения проявляются гипергидрозом, тахикардией, лабильностью пульса).
Ряд авторов выделяет третью форму Г. б., развивающуюся в юношеском возрасте или у взрослых и характеризующуюся преимущественным поражением ц. н. с., но имеющую более мягкое течение, сходное с хрон, формой.
Рентгенологическая картина. Рентгенол, изменения обнаруживают в костной системе, легких и в редких случаях в жел.-киш. тракте, гл. обр. при хрон, форме болезни.
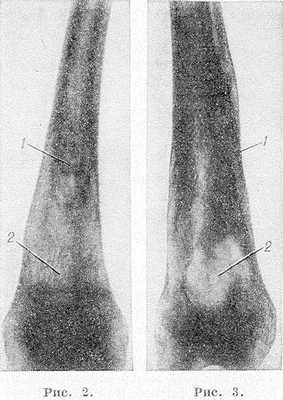
Рис. 2 и 3. Рентгенограммы бедер при болезни Гоше. Рис. 2. Булавовидное расширение метадиафиза бедренной кости в нижней трети с выраженными внутрикостными обызвествлениями (1) и широкопетлистой костной структурой (2). Рис.З. Расширение метадиафиза бедренной кости с уплотнением (1) и обширным участком деструкции (2).
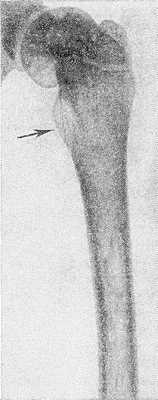
Рис. 4. Рентгенограмма бедра ребенка 10 лет при болезни Гоше: выражено утолщение шейки бедренной кости (указано стрелкой).
Наиболее часто изменения обнаруживают в длинных трубчатых костях и позвоночнике. Первое место по частоте поражения занимает дистальная половина бедра, где выявляют характерное веретенообразное или булавовидное вздутие кости (рис. 2 и 3) с истончением коркового слоя, к-рое часто сочетается с грубоячеистой структурой, внутрикостными обызвествлениями, отдельными очагами деструкции и иногда с эностальным склерозом кости. Периостозы, как правило, не развиваются; иногда могут наблюдаться обширные краевые деструкции, сопровождающиеся периостозами. В головке бедренной кости образуются асептические некрозы по типу болезни Пертеса со свойственной данному заболеванию стадийностью процесса и исходами (см. Пертеса болезнь). Может наблюдаться утолщение шейки бедра (рис. 4) с образованием coxa vara.
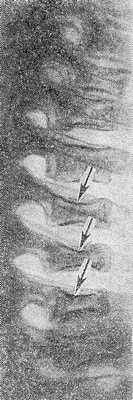
Рис. 5. Рентгенограмма грудного отдела позвоночника ребенка 10 лет при болезни Гоше: выражена бревиплатиспондилия (уплощение тел позвонков — указано стрелками).
Патологические переломы тел позвонков могут привести к клиновидной их деформации. Сохранность межпозвоночного диска позволяет исключить туберкулезный характер поражения. В ряде случаев развивается множественное поражение тел позвонков, сопровождающееся уменьшением их высоты и приводящее к снижению роста больного — системная бревиплатиспондилия (рис. 5).
Изменения в органах грудной полости выявляются редко. В легких рентгенологически отмечают усиление интерстициального рисунка с точечными очажками уплотнения, иногда в сочетании с увеличением внутригрудных лимфатических узлов. Еще более редки поражения жел.-киш. тракта, характеризующиеся появлением в желудке дефектов наполнения.
Диагноз
Диагноз ставят на основании клинической картины, данных лабораторных и рентгенологических исследований. Основным критерием для диагностики Гоше болезни является обнаружение в пунктате селезенки, костного мозга или печени клеток Гоше. В пунктате наряду с отдельно лежащими клетками могут встречаться и синцитиальные образования из них. Описаны единичные случаи вымывания клеток Гоше в периферическую кровь и выявления их методом лейкоконцентрации (см. Лейкоциты).
Г. б. можно диагностировать биохим. методом, выявляя дефицит фермента глюкоцереброзидазы в лейкоцитах периферической крови, в культуре фибробластов кожи и в культуре клеток из амниотической жидкости. У больных с острой формой Г. б. содержание фермента составляет 5—10% нормы, у больных с хрон, формой — 40—60%.
Для диагностики Г. б. определяют также содержание глюкоцереброзидов в осадке мочи больного или в биопсийном материале, напр, в печеночной ткани. Содержание этих глюколипидов повышено. В сыворотке крови больного обнаруживается избыток кислой фосфатазы, выявляется гиперкальциемия.
Исследование культуры клеток из амниотической жидкости позволяет выявлять носителей дефицита фермента глюкоцереброзидазы в пренатальном периоде жизни.
Лечение
Лечение острой формы симптоматическое. Основной метод лечения хрон. формы Г. б.— спленэктомия (см.). Попытка терапии отдельных форм сфинголипидозов, к к-рым относится Г. б., введением в плазму больных недостающих ферментов пока не дала надежных результатов.
Прогноз при острой форме Гоше болезни неблагоприятный; смерть наступает на 1—2-м году жизни. Прогноз хрон, формы, как правило, благоприятный.
Профилактика
В случае рождения в семье ребенка со злокачественной формой болезни при последующих беременностях матери больного показано исследование амниотической жидкости плода. При диагностике Гоше болезни у 11—17-недельного плода показано прерывание беременности. Специфической профилактики нет.
Библиография: Берестов А. И. и Ковригин А. Е. К биохимической и цитохимической характеристике болезни Гоше у детей, Педиатрия, № 8, с. 33, 1972, библиогр.; Гусев Е.И. Клиническое и биохимическое изучение наследственных болезней обмена веществ с поражением нервной системы, Журн, невропат, и психиат., т. 71, № 10, с. 1475, 1971; Егоров П. И. и Мищенко А. С. Клиника, диагностика и лечение болезни Гоше, Педиатрия, № 8, с. 53, 1969, библиогр.; Жарко К. П., Митасова И. Н. и Ермакова Р.П. Болезнь Гоше у взрослых, Врач, дело, № 6, с. 143, 1969;
Покровский П. И. и Цепа Л. С. К вопросу о клинике, диагностике и терапии болезни Гоше, Пробл, гематол, и перелив, крови, т. 16, № И, с. 15, 1971, библиогр.; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, т. 1, с. 502, М., 1964; Bradу R. О., Jоhnsоn W. G. a. Uhlendorf B. W. Identification of heterosygous carriers of lipid storage disease, Amer. J. Med., v. 51, p. 423, 1971, bibliogr.; Brady R. O., KanferJ. N. a. Shapiro D. Metabolism of glucocerebrosides, Biochem. biophys. Res. Commun., v. 18, p. 221, 1965; Brooks S. E.H. a. Audretsch J. J. Ultrastructural diagnosis of Gaucher’s disease with negative-staining technique, Arch. Path., v. 95, p. 226, 1973; Cerebral sphingolipidoses, ed. by S. M. Aronson a. B. W. Volk, p. 73, N. Y. — L., 1962, bibliogr.; Danz M. u. Ka tenkamp D. Zur Gehirnbeteiligung beim Kongenitalen und friihinfantilen Morbus Gaucher, Zbl. allg. Path., Bd 115, S. 536, 1972; Gaucher P. E. De l’6pith61ioma primitif et isol6 de la rate, P., 1882; Goll E. У. u. Pexa H. Uber andauernde Ausschwemmung von Gaucher-Zellen ins Blut, Acta haemat. (Basel), Bd 31, S. 113,1964; Katz M. a. o. Maladie de Gaucher, J. Radiol. Electrol., t. 54, p. 61, 1973; The metabolic basis inherited disease, ed. by J. B. Stanbury a. o., p. 730, N. Y., 1972, bibliogr.; Miller J. D. Gaucher’s disease, Ann. intern. Med., v. 78, p. 883, 1973; Rosenfld-Striсhar d N. G. La maladie de Gaucher, 6tude d’une forme familiale, P., 1965, bibliogr.; Schneider E.L. a.o. Infantile (typ II) Gaucher’s disease, J. Pediat., v. 81, p. 1134, 1972, bibliogr.
М. Д. Бриллиант, А. И. Воробьев; E. И. Гусев (невр.), Э. 3. Новикова (рент.).
Болезнь Гоше
Болезнь Гоше - это генетическое заболевание, характеризующееся нарушением липидного обмена, недостаточностью лизосомальных ферментов, накоплением гликолипидов в клеточных структурах. Симптомы определяются типом патологии. Общими признаками являются увеличение печени, селезенки, снижение свертываемости крови. При I типе выявляются нарушения со стороны костной системы: остеопороз, частые переломы, инфекции костей. При II и III типе доминирует неврологическая симптоматика: судороги, паралич, косоглазие, задержка умственного развития. Диагностика основана на биохимическом анализе дефицитарного фермента. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
МКБ-10
Общие сведения
Заболевание получило свое название по фамилии французского врача Филиппа Гоше. В 1882 году он описал симптомы и патанатомические особенности строения селезенки пациентки, которая умерла от сепсиса. Спустя несколько десятилетий при аналогичном клиническом случае Гоше определил накопление в селезенке глюкоцереброзида и недостаточность фермента глюкоцереброзидазы. Болезнь Гоше (сфинголипидоз, глюкозилцерамидный липидоз) относится к группе лизосомальных болезней накопления - наследственных патологий, при которых изменены функции клеточных органелл лизосом. Частота заболевания составляет от 1:40 тыс. до 1:70 тыс. Распространенность наиболее велика в сообществах, где допустимы браки между близкими родственниками, например, у евреев ашкенази. Носительство мутационного гена определяется примерно у 1 человека из 400.
Причины
Глюкозилцерамидный сфинголипидоз является наиболее частой формой наследственных ферментопатий. Причиной его развития считается дефект гена GBA, который кодирует фермент лизосом бета-глюкозидазу (глюкоцереброзидазу), ответственную за расщепление липидов. Наследование болезни происходит аутосомно-рецессивным способом, для формирования ферментопатии необходимо присутствие пары измененных генов: один - от матери, другой - от отца. В супружеской паре, где оба родителя - носители мутации, вероятность рождения больного ребенка составляет 25%. Риск передачи одного дефектного гена, то есть риск носительства без развития болезни в таких семьях равен 50%. При наличии в генотипе двух мутантных аллелей функция глюкоцереброзидазы снижается на 15-30% от нормального уровня.
Патогенез
Патогенетической основой болезни является снижение каталитической активности бета-глюкозидазы. В результате нарушается процесс расщепления гликосфинголипидов (сложных соединений липидов и углеводов) до глюкозы и церамида. Аномально прогрессивное накопление макромолекул происходит в клетках, которые характеризуются повышенной скоростью их обновления - в макрофагах. Негидролизованные липиды концентрируются в лизосомах, образуются особые клетки накопления - клетки Гоше. Первичный метаболический сбой провоцирует вторичные расстройства биохимических процессов и клеточных функций. Из-за патологии жирового обмена развивается синдром активации макрофагов. Стимулируется моноцитопоэз, увеличивается содержание макрофагов в печени, селезенке, костном мозге. Это становится причиной спленомегалии, гепатомегалии, инфильтрации костного мозга. Расстройство регуляторной функции макрофагов является провоцирующим фактором цитопении, поражения костей и суставов.
Симптомы болезни Гоше
По возрасту дебюта и особенностям клинической картины выделяют три типа болезни. Первый тип наиболее распространен, имеет хронический характер течения. Симптомы чаще проявляются к 30-40 годам, реже болезнь манифестирует в детском возрасте. Увеличение размеров печени и селезенки начинается сразу после рождения, но клинически проявляется позже. Первыми признаками патологии становятся анемия, повышенная кровоточивость. Угнетение системы кроветворения сопровождается снижением уровня гемоглобина и тромбоцитов. Изменения со стороны опорно-двигательного аппарата представлены болями в костях и суставах, частыми переломами, деформациями (как правило, изменяется бедренная кость). У взрослых заметна гиперпигментация на лице и ногах: кожа темнеет, приобретает оттенок от желтоватого до желто-коричневого. Возможно появление плоских красных пятен с типичной локализацией в области вокруг глаз. Рост пациентов ниже среднего.
Второй тип болезни (острый инфантильный или острый нейропатический) встречается очень редко, развивается в промежутке от рождения до полутора лет, чаще всего симптомы дебютируют в первые три месяца жизни. Характеризуется стремительным течением, плохим откликом на лечение. На первый план выходят неврологические расстройства, спровоцированные скоплением клеток Гоше в центральной нервной системе. Дети слабо кричат, вяло сосут. Нарушен глотательный рефлекс, нередко отмечаются сбои цикла дыхания. Наблюдается заметная задержка психического и физического развития. На начальной стадии заболевания мышечный тонус снижен, через 9-12 месяцев после дебюта возникает гипертонус, особенно в мышцах шеи и конечностях. Развиваются судороги, косоглазие, спастический паралич. Печень и селезенка увеличены. Дети часто болеют тяжелой пневмонией.
Третий тип - ювенильный или подострый нейропатический. Первые признаки - увеличение селезенки и печени - возникают в 2-3 года. Полная симптоматика разворачивается в период с 6 до 15 лет. Клинические проявления поражения ЦНС включают гипертонус мышц, паралич спастического типа, косоглазие, непроизвольные спазмы, судороги, затрудненный цикл дыхания с трудностью вдоха, проблемы при глотании. Имеются расстройства психического развития: снижение интеллектуальных функций, несформированность речи и письма, эмоциональная неустойчивость, психозы. Дети отстают в половом развитии. Течение болезни неуклонно прогрессирующее.
Осложнения
Наиболее тяжелые осложнения выявляются при втором и третьем типе болезни. Поражение спинного и головного мозга приводит к нарушению дыхательного цикла, развиваются внезапные остановки дыхания, возрастает риск спазма гортани и смерти от удушья. Сниженный уровень тромбоцитов способен стать причиной обширных внутренних кровотечений. У больных с патологией первого типа распространенным осложнением является разрушение костей, их повышенная ломкость и инфекционные поражения. Ограничивается подвижность, пациенты не могут передвигаться самостоятельно, нуждаются в постороннем уходе.
Диагностика
Сбор анамнеза и физикальное обследование выполняется врачом-эндокринологом и неврологом, дополнительно назначаются консультации генетика, гематолога, офтальмолога, педиатра, психиатра. Анамнестические данные включают наличие болезни Гоше у родственников. При осмотре выявляются типичные признаки: низкий рост, патологии костей, неврологические симптомы (косоглазие, атаксия, паралич), геморрагический синдром, гиперпигментация кожи. Иногда подозрение на заболевание возникает после случайного выявления увеличенной селезенки на снимках УЗИ, угнетении кроветворной системы по данным общего анализа крови. Для подтверждения диагноза, исключения других метаболических наследственных патологий, остеомиелита, костного туберкулеза, вирусного гепатита и онкологических поражений крови проводится специфическая диагностика:
- Клиническое, биохимическое исследование крови. У большинства больных определяется тромбоцитопения, лейкопения, анемия, которая у детей обычно имеет железодефицитное происхождение. В результатах биохимического анализа обнаруживается сниженная активность глюкоцереброзидазы.
- Ферментный анализ клеток. При болезни Гоше в образцах сухой крови и в фибробластах кожи выявляется недостаточная активность глюкозидазы. Степень дефицитарности фермента не имеет прямой корреляции с выраженностью симптомов. Дополнительный биохимический маркер - хитотриозидаза. Этот фермент синтезируется активированными макрофагами, характерно повышение его активности в 6-10 раз.
- Морфологическое изучение костного мозга. Подтверждается наличие специфических для данного заболевания структур - клеток Гоше. Результат позволяет исключить гемобластоз и лимфопролиферативное заболевание.
- Исследование структуры костной ткани. С целью оценки тяжести поражения костно-суставной системы выполняется денситометрия, рентгенография и/или МРТ костей скелета. Возможен диффузный остеопороз, могут визуализироваться колбы Эрленмейера, очаги остеолизиса, остеосклероза и остеонекроза. На ранних стадиях болезни отмечается остеопения, инфильтрация костного мозга.
- Визуализирующее исследование селезенки, печени. Проводится УЗИ и МРТ внутренних органов. По результатам устанавливается наличие или отсутствие очаговых поражений, измеряется объем увеличенного органа. Исходные показатели в последующем позволяют контролировать эффективность терапии.
- Молекулярно-генетические исследования. ДНК-диагностика является необязательной процедурой. Подтверждение мутации в гене GBA бывает необходимо при неоднозначности биохимических исследований, а также в рамках пренатальных и преимплантационных обследований.
Лечение болезни Гоше
Специализированная помощь больным с первым и третьим типом болезни направлена на устранение симптомов и компенсацию первичного генетического дефекта - увеличение количества недостающего фермента, усиление катаболизма гликосфинголипидов. При 2 типе патологии терапевтические мероприятия оказываются недостаточно эффективными, усилия врачей сводятся к облегчению клинических проявлений - болей, судорог, дыхательных расстройств. Общая схема включает следующие направления:
- Ферментозаместительная терапия. Основным методом лечения является пожизненная ферментная заместительная терапия (ФЗТ) с применением рекомбинантной глюкоцереброзидазы. Эффективность достаточно высока - симптомы полностью купируются, качество жизни больных повышается. ФЗТ целесообразна при третьем и первом типе заболевания. Препараты вводятся внутривенно. Частые инфузии иногда становятся причиной воспалительных заболеваний вен (флебитов).
- Субстрат-редуцирующая терапия. Данное направление является новым в лечении болезни Гоше, относительно широко распространено в США и странах Европы. Нацелено на снижение скорости производства субстрата гликосфинголипидов и ускорение катаболизма накапливающихся макромолекул. В качестве препаратов выступают специфические ингибиторы глюкозилцерамидсинтазы. Метод показан при заболевании 1 типа с легкими и умеренными симптомами.
- Симптоматическая терапия. При явлениях остеопороза назначается комплексная терапия, включающая прием кальцийсодержащих препаратов, витамина D и соблюдение диеты, обогащенной кальцием. Эти меры позволяют замедлить потерю костной массы, повысить прочность костей, предотвратить переломы. При скелетных осложнениях применяются анальгезирующие средства (НПВС), антибактериальная терапия. Симптомы неврологических нарушений купируются противоэпилептическими препаратами, ноотропами, миорелаксантами.
Прогноз и профилактика
Благоприятный исход наиболее вероятен у пациентов с 1 типом заболевания - комплексный терапевтический подход позволяет нормализовать функциональность глюкоцереброзидазы, предупредить развитие осложнений, избежать инвалидизации. При 3 типе прогноз зависит от характера течения болезни, индивидуальной реакции организма на лечебные мероприятия. 2 тип имеет крайне тяжелые проявления и завершается гибелью больного. Профилактика проводится во время планирования беременности и на ее начальных сроках. Медико-генетическое консультирование рекомендуется семьям, имеющим близких родственников с данной патологией. При высоком риске передачи мутации будущему ребенку в первом триместре выполняется исследование уровня фермента в амниотической жидкости, решается вопрос о прерывании беременности.
2. Болезнь Гоше: современная диагностика и лечение/ Лукина Е.А.// Клиническая онкогематология. - 2009 - Т.2, №2.
4. Болезнь Гоше: клиническая картина, диагностика, лечение/ Давыдкин И.Л., Хайретдинов Р.К., Кривова С.П., Данилова О.Е.// Эффективная фармакотерапия. - 2014 - №47.
Болезнь Гоше у детей
Болезнь Гоше (БГ) - наиболее частая форма наследственных ферментопатий, объединенных в группу лизосомных болезней накопления, в основе которой лежит дефект гена GBA, кодирующего лизосомный фермент β-D-глюкозидазу (глюкоцереброзидазу), ответственный за катаболизм липидов.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
• Болезнь Гоше, I тип. Патологический перелом шейки правого бедра, состояние после оперативного лечения.
При II и III типах БГ в патологический процесс вовлекается нервная система, поэтому их называют нейронопатическими [2,3,4,5,6].
БГ наследуется по аутосомно-рецессивному типу. Присутствие двух мутантных аллелей гена GBA ассоциируется со значительным ( ≤ 30% от нормального уровня) снижением каталитической активности глюкоцереброзидазы, функция которой заключается в деградации гликосфинголипидов (или глюкоцереброзидов, глюкозилцерамидов) до глюкозы и церамидов.
Дефицит фермента приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше).
Следствием данного метаболического дефекта являются: хроническая активация макрофагальной системы, аутокринная стимуляция моноцитопоэза и увеличение абсолютного количества макрофагов, нарушение регуляторных функций макрофагов. Ген GBA, кодирующий глюкоцереброзидазу, расположен в хромосомной области 1q21. В настоящее время описано около 400 различных мутаций, патогенность которых проявляется широким полиморфизмом клинических симптомов, обусловленных частичной или полной потерей каталитической активности кодируемого фермента [1,2,3,4,5,6].
Эпидемиология
Частота БГ составляет 1:40000 - 1:70000. В популяции евреев-ашкенази (выходцев из Восточной Европы) частота встречаемости этого заболевания является более высокой и достигает 1:450 - 1:1000 [6].
• проявлений спонтанного геморрагического синдрома (в виде подкожных гематом, кровоточивости слизистых оболочек) или длительных кровотечений при малых оперативных вмешательствах;
• болей в костях и суставах; давность, характер и локализацию болей, наличие в прошлом переломов костей;
• неврологической симптоматики (глазодвигательная апраксия или сходящееся косоглазие, атаксия, потеря интеллекта, нарушения чувствительности и др.);
• семейного анамнеза (наличие спленэктомии или перечисленных выше симптомов у родных братьев и сестер [1,2,3,4,5,6]).
• При проведении клинического осмотра рекомендуется включать измерение роста и массы тела, температуры тела, оценку состояния костно-суставной системы; выявление признаков геморрагического синдрома, гепатоспленомегалии, лимфаденопатии, а также своеобразной гиперпигментации кожных покровов в области коленных и локтевых суставов, характерной для пациентов с БГ [1,2,3,6].
Комментарии: Клинические проявления БГ I типа разнообразны, а возраст манифестации варьирует. БГ типа 1 имеет хроническое течение. Клиническая картина характеризуется прогрессирующим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и патологией трубчатых костей скелета (болезненными костными кризами и аваскулярными некрозами эпифизов, чаще головки бедренной кости).
Основные симптомы заболевания при БГ II типа возникают в первые 6 мес жизни (острая) нейропатическая форма с бульбарной и пирамидной симптоматикой, когнитивной задержкой. Течение заболевания - быстро прогрессирующее. Клинический симптомокомплекс включает признаки поражения нервной системы и внутренних органов:
• тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы;
Главной особенностью клинических проявлений БГ III типа является то, что наряду с поражением паренхиматозных органов (гепатоспленомегалия) наблюдаются неврологические проявления, сходные с таковыми при типе 2 БГ, но менее тяжело выраженные и возникающие, как правило, в возрасте от 6 до 15 лет и позже:
Комментарии: у большинства больных с БГ выявляет тромбоцитопению, лейкопению и анемию, как проявления гиперспленизма.
Комментарии: морфологическое исследование костного мозга способствует выявлению характерных диагностических элементов - клеток Гоше и одновременно исключению диагноза гемобластоза или лимфопролиферативного заболевания как причины цитопении и гепатоспленомегалии. Детям это исследование проводят редко, строго по показаниям.
• Рекомендуется проведение биохимического исследования: определение активности β-D-глюкозидазы в лейкоцитах периферической крови, пятнах крови, высушенной на фильтровальной бумаге, определение активности хитотриозидазы в плазме крови [1,2,3,4,6].
• Рекомендовано проведение молекулярно-генетического исследования: выявление мутаций в гене GBА методом секвенирования кодирующих и прилегающих интронных областей [6].
• Рекомендуется оценить уровень сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18 [6].
Комментарии. Характерными лабораторными симптомами при БГ также являются: повышение уровня сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18, которые отражают степень активности заболевания и могут использоваться как биомаркеры для оценки динамики на фоне лечения.
Комментарии: рентгенография костей скелета необходима для выявления и оценки тяжести поражения костно-суставной системы. Изменения костной ткани могут быть представлены диффузным остеопорозом, характерной колбообразной деформацией дистальных отделов бедренных и проксимальных отделов большеберцовых костей (колбы Эрленмейера), очагами остеолизиса, остеосклероза и остеонекроза, патологическими переломами.
Комментарии: денситометрия и магнитно-резонансная томография (МРТ являются более чувствительными методами и позволяют диагностировать поражение костей (остеопению, инфильтрацию костного мозга) на ранних стадиях, не доступных визуализации рентгенографией.
Комментарии: УЗИ и МРТ печени и селезенки позволяют выявить их очаговые поражения и определить исходный объем органов, что необходимо для последующего контроля эффективности заместительной ферментной терапии.
• Рекомендовано проведение эзофагогастродуоденоскопия при наличии соответствующих жалоб или признаков портальной гипертензии [3,5,6].
• Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска [1,2,3,4,5,6].
Комментарии: как и при других аутосомно-рецессивных заболеваниях, при БГ для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11-й неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22-й неделе беременности.
Дифференциальный диагноз
Диагноз болезнь Гоше ставится на основании совокупности клинических данных, результатов лабораторного исследования, биохимического и молекулярно-генетического анализа.
Диагноз БГ следует заподозрить у пациента с необъяснимой сплено- и гепатомегалией, цитопенией и симптомами поражения костей.
Золотым стандартом диагностики является биохимическое тестирование, поскольку патогенность некоторых выявленных редких и новых мутаций требует дополнительных доказательств [2,6].
Для I типа БГ в зависимости от вида манифестации - разнообразные экзогенные и наследственные заболевания, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью (вирусные гепатиты, остеомиелит, костный туберкулез, гемофилия, гликогеноз, болезнь Нимана-Пика (тип В), недостаточность кислой липазы).
Для II и III типов БГ - болезнь Нимана-Пика (типы А, С), GM1-ганглиозидоз, галактосиалидоз, дефицит лизосомной кислой липазы, а также врожденная окуломоторная апраксия.
• Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с подтвержденным диагнозом БГ I
• Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с хроническим поражением нервной системы (БГ III тип), у которых имеются клинически значимые неневрологические проявления заболевания [2,4,5,6].
• Не рекомендована ферментная заместительная терапия при II типе БГ, так как не эффективна [1,2,3,5,6].
Комментарии: ФЗТ обеспечивает устойчивое улучшение состояния пациентов: нормализует уровни гемоглобина, тромбоцитов; объем печени и селезенки (у неспленэктомизированных больных); купирует костные боли, предотвращает развитие остеонекрозов и костных кризов; приводит к нормализации роста и значительно повышает качество жизни детей с болезнью Гоше.
- велаглюцераза альфа ж,7н .
Имиглюцераза ж,7н - модифицированная форма кислой β-глюкозидазы человека, у детей с БГ начальная доза имиглюцеразы составляет:
- при I типе БГ без поражения трубчатых костей скелета - 30 ЕД/кг на 1 введение [6,7,8];
Велаглюцераза альфа ж,7н показана для длительного лечения детей с болезнью Гоше I типа. Рекомендуемая доза составляет 30-60 ЕД/кг 1 раз в 2 недели.
Дозу можно корректировать индивидуально, на основании достижения ожидаемого эффекта и его сохранения, однако применение доз выше 60 ЕД/кг не изучено [6,12,13,14,15].
При обследовании сиблингов (братьев и сестер пробанда) могут быть выявлены дети с БГ, не имеющие клинических проявлений. Такие пациенты нуждаются в наблюдении, начинать их лечение необходимо при появлении первых симптомов болезни.
• При развитии проявлений остеопороза для замедления и прекращения потери костной массы, повышения ее прочности, предотвращения переломов костей в комплексной терапии рекомендуется применять: альфакальцидолж,вк, соли кальцияж,вк [1,3,5,6]
Комментарии: в качестве симптоматической терапии скелетных осложнений БГ I типа назначаются анальгетики (во время костных кризов), нестероидные противовоспалительные средства, редко, лишь при наличии показаний, - антибактериальная терапия.
• При доказанном диагнозе БГ не рекомендованы повторные пункции костного мозга и другие инвазивные диагностические мероприятия (биопсия печени, селезенки) [1,3,5,6].
• Не рекомендовано оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования [1,2,5,6].
• Противопоказано назначение кортикостероидов с целью купирования цитопенического синдрома [1,2,5,6].
• Не рекомендовано и не обосновано назначение препаратов железа больным с развернутой картиной БГ, так как анемия в этих случаях носит характер «анемии воспаления» [1,2,5,6].
(Сила рекомендаций - 1; достоверность доказательств - С)
Ведение пациентов рекомендуется осуществлять в соответствии с рекомендациями по минимально необходимому мониторингу состояния больных при БГ I типа, разработанными Объединенной международной группой по изучению болезни Гоше (International Collaborative Gaucher Group). Контроль показателей крови необходимо проводить 1 раз в 3 мес, размеров паренхиматозных органов (УЗИ, МРТ) - 1 раз в 6 мес, а также при изменении дозировки препарата или при значительных клинических осложнениях (табл. 1). Определение состояния костной ткани осуществляют 1 раз в год. Определение активности хитотриазидазы на фоне ферментной заместительной терапии проводят 1 раз в 12 мес.
Рекомендуется пациентов с БГ наблюдать по месту жительства в амбулаторно-поликлинических условиях врачам педиатрам, гематологам, при БГ III типа - неврологам, при наличии костных нарушений - ортопедам, до достижения возраста 18 лет.
Рекомендуется введение ФЗТ проводить регулярно при наличии показаний в случае осложненного течения болезни - в условиях круглосуточного стационара, в стабильном состоянии - в стационаре дневного пребывания или амбулаторно 1 раз в 2 недели. До достижения клинико-лабораторной ремиссии все пациенты с БГ должны проходить контрольное обследование с целью коррекции дозы ФЗТ в круглосуточном или дневном стационаре 2 раза в год; в дальнейшем обследование проводится 1 раз в год.
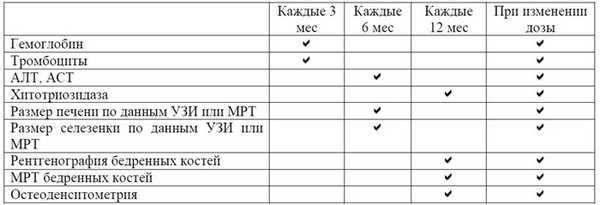
Таблица 1- Принципы мониторинга болезни Гоше
АЛТ/АСТ ― аланин-/аспартатаминотрансферазы, УЗИ ― ультразвуковое исследование, МРТ ― магнитно-резонансная томография.
Болезнь Гоше у детей. Клинические рекомендации.
Ферментная заместительная терапия - лечение, заключающееся в пожизненном введении препарата (рекомбинантного энзима) пациентам с врожденным дефектом метаболизма.
1. Краткая информация
1.1 Определение
Болезнь Гоше (БГ) - наиболее частая форма наследственных ферментопатий, объединенных в группу лизосомных болезней накопления, в основе которой лежит дефект гена GBA, кодирующего лизосомный фермент ?-D-глюкозидазу (глюкоцереброзидазу), ответственный за катаболизм липидов.
1.2 Этиология и патогенез
БГ наследуется по аутосомно-рецессивному типу. Присутствие двух мутантных аллелей гена GBA ассоциируется со значительным (? 30% от нормального уровня) снижением каталитической активности глюкоцереброзидазы, функция которой заключается в деградации гликосфинголипидов (или глюкоцереброзидов, глюкозилцерамидов) до глюкозы и церамидов. Дефицит фермента приводит к накоплению в лизосомах макрофагов неутилизированных липидов и образованию характерных клеток накопления (клеток Гоше). Следствием данного метаболического дефекта являются: хроническая активация макрофагальной системы, аутокринная стимуляция моноцитопоэза и увеличение абсолютного количества макрофагов, нарушение регуляторных функций макрофагов. Ген GBA, кодирующий глюкоцереброзидазу, расположен в хромосомной области 1q21. В настоящее время описано около 400 различных мутаций, патогенность которых проявляется широким полиморфизмом клинических симптомов, обусловленных частичной или полной потерей каталитической активности кодируемого фермента [1,2,3,4,5,6].
1.3 Эпидемиология
1.4 Кодирование по МКБ-10
Е75.2 - Другие сфинголипидозы
1.5 Примеры диагнозов
- Болезнь Гоше, I тип.
- Болезнь Гоше, II тип.
- Болезнь Гоше, III тип.
- Болезнь Гоше, I тип. Состояние после спленэктомии.
- Болезнь Гоше, I тип. Патологический перелом шейки правого бедра, состояние после оперативного лечения.
- Болезнь Гоше, I тип. Асептический некроз головки бедренной кости слева.
- Болезнь Гоше, I тип. Остеопения поясничного отдела позвоночника.
- Болезнь Гоше, III тип. Симптоматическая эпилепсия.
- Болезнь Гоше, III тип. Косоглазие содружественное сходящееся альтернирующее.
1.6 Классификация
В зависимости от клинического течения выделяют 3 типа болезни Гоше:
- I тип - ненейронопатический (самый частый);
- II тип - инфантильный или острый нейронопатический;
- III тип - подострый нейронопатический.
2. Диагностика
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб следует обратить внимание на наличие:
- задержки физического и полового развития;
- слабости, повышенной утомляемости, частых респираторных инфекций;
- проявлений спонтанного геморрагического синдрома (в виде подкожных гематом, кровоточивости слизистых оболочек) или длительных кровотечений при малых оперативных вмешательствах;
- болей в костях и суставах; давность, характер и локализацию болей, наличие в прошлом переломов костей;
- предшествующей спленэктомии (полной или частичной);
- неврологической симптоматики (глазодвигательная апраксия или сходящееся косоглазие, атаксия, потеря интеллекта, нарушения чувствительности и др.);
- семейного анамнеза (наличие спленэктомии или перечисленных выше симптомов у родных братьев и сестер [1,2,3,4,5,6]).
2.2 Физикальное обследование
- При проведении клинического осмотра рекомендуется включать измерение роста и массы тела, температуры тела, оценку состояния костно-суставной системы; выявление признаков геморрагического синдрома, гепатоспленомегалии, лимфаденопатии, а также своеобразной гиперпигментации кожных покровов в области коленных и локтевых суставов, характерной для пациентов с БГ [1,2,3,6].
Комментарии: Клинические проявления БГ I типа разнообразны, а возраст манифестации варьирует. БГ типа 1 имеет хроническое течение. Клиническая картина характеризуется прогрессирующим увеличением паренхиматозных органов (печени и селезенки), панцитопенией и патологией трубчатых костей скелета (болезненными костными кризами и аваскулярными некрозами эпифизов, чаще головки бедренной кости).
Основные клинические симптомы БГ I типа:
- гепатоспленомегалия,
- геморрагический синдром,
- костные боли (костные кризы),
- нарушение подвижности в суставах, обусловленное асептическим некрозом,
- патологические переломы,
- задержка физического и полового развития,
- астенический синдром.
Основные симптомы заболевания при БГ II типа возникают в первые 6 мес жизни (острая) нейропатическая форма с бульбарной и пирамидной симптоматикой, когнитивной задержкой. Течение заболевания - быстро прогрессирующее. Клинический симптомокомплекс включает признаки поражения нервной системы и внутренних органов:
- гепатоспленомегалия;
- нарушение глотания, поперхивание, часто осложняющиеся аспирационной пневмонией;
- тризм, билатеральное фиксированное косоглазие, прогрессирующая спастичность с ретракцией шеи, гиперрефлексия, положительный симптом Бабинского и другие патологические рефлексы;
- прогрессирующая задержка психомоторного развития и потеря ранее приобретенных навыков;
- тонико-клонические судорожные приступы, резистентные к противосудорожной терапии.
Главной особенностью клинических проявлений БГ III типа является то, что наряду с поражением паренхиматозных органов (гепатоспленомегалия) наблюдаются неврологические проявления, сходные с таковыми при типе 2 БГ, но менее тяжело выраженные и возникающие, как правило, в возрасте от 6 до 15 лет и позже:
- окуломоторные расстройства;
- снижение интеллекта (от незначительных изменений до тяжелой деменции);
- экстрапирамидная ригидность;
- мозжечковые нарушения;
- расстройства речи, письма;
- поведенческие изменения, эпизоды психоза;
- миоклонии, генерализованные тонико-клонические судороги.
В большинстве случаев течение заболевания медленно прогрессирующее [1,2,3,6].
2.3 Лабораторная диагностика
- Рекомендуется проведение клинического анализа крови [1,2,3,4,6].
Комментарии: у большинства больных с БГ выявляет тромбоцитопению, лейкопению и анемию, как проявления гиперспленизма.
- Рекомендовано морфологическое исследование костного мозга [1,2,3,4,6].
- Рекомендуется проведение биохимического исследования: определение активности ?-D-глюкозидазы в лейкоцитах периферической крови, пятнах крови, высушенной на фильтровальной бумаге, определение активности хитотриозидазы в плазме крови [1,2,3,4,6].
- Рекомендовано проведение молекулярно-генетического исследования: выявление мутаций в гене GBА методом секвенирования кодирующих и прилегающих интронных областей [6].
- Рекомендуется оценить уровень сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18 [6].
Комментарии. Характерными лабораторными симптомами при БГ также являются: повышение уровня сывороточного ферритина, ангиотензинпревращающего фермента, хемокина CCL 18, которые отражают степень активности заболевания и могут использоваться как биомаркеры для оценки динамики на фоне лечения.
2.4 Инструментальная диагностика
- Рекомендовано проведение рентгенографии костей скелета [1,2,3,4,6].
- Рекомендовано проведение денситометрии и магнитно-резонансной томографии (МРТ) [1,2,3,4,6].
- Рекомендовано проведение ультразвукового исследования (УЗИ) и МРТ печени и селезенки [1,2,3,4,6].
- Рекомендовано проведение допплер-эхокардиографии у спленэктомированных пациентов [3,5,6].
- Рекомендовано проведение эзофагогастродуоденоскопия при наличии соответствующих жалоб или признаков портальной гипертензии [3,5,6].
2.4 Иная диагностика
Консультации специалистов пациентам с подозрением на болезнь Гоше рекомендуются по показаниям.
- Рекомендована консультации психоневролога [1,2,3,4,5,6]
Комментарии: необходима всем детям с БГ для уточнения типа заболевания.
Комментарии: показана при подозрении на наличие у ребенка скелетной патологии.
- Рекомендована консультация врача-генетика [1,2,3,4,5,6].
Комментарии: показана семьям, имеющим родственников с болезнью Гоше.
2.5. Медико-генетическое консультирование и пренатальная диагностика
- Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска [1,2,3,4,5,6].
Комментарии: как и при других аутосомно-рецессивных заболеваниях, при БГ для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики.
Пренатальная диагностика проводится молекулярно-генетическими или биохимическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9-11-й неделе беременности и/или клеток амниотической жидкости, плодной крови на 20-22-й неделе беременности.
2.6 Дифференциальная диагностика
Диагноз болезнь Гоше ставится на основании совокупности клинических данных, результатов лабораторного исследования, биохимического и молекулярно-генетического анализа.
Диагноз БГ следует заподозрить у пациента с необъяснимой сплено- и гепатомегалией, цитопенией и симптомами поражения костей.
Золотым стандартом диагностики является биохимическое тестирование, поскольку патогенность некоторых выявленных редких и новых мутаций требует дополнительных доказательств [2,6].
Для I типа БГ в зависимости от вида манифестации - разнообразные экзогенные и наследственные заболевания, сопровождающиеся висцеромегалией, острыми болями в костях, кровоточивостью (вирусные гепатиты, остеомиелит, костный туберкулез, гемофилия, гликогеноз, болезнь Нимана-Пика (тип В), недостаточность кислой липазы).
Для II и III типов БГ - болезнь Нимана-Пика (типы А, С), GM1-ганглиозидоз, галактосиалидоз, дефицит лизосомной кислой липазы, а также врожденная окуломоторная апраксия.
3. Лечение
3.1 Консервативное лечение
- Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с подтвержденным диагнозом БГ I типа без поражения нервной системы [1,2,3,5,6].
(Сила рекомендаций - 1; достоверность доказательств - В)
- Рекомендована пожизненная ферментная заместительная терапия (ФЗТ) рекомбинантной глюкоцереброзидазой пациентам с хроническим поражением нервной системы (БГ III тип), у которых имеются клинически значимые неневрологические проявления заболевания [2,4,5,6].
(Сила рекомендаций - 2; достоверность доказательств - В)
Комментарии: ФЗТ обеспечивает устойчивое улучшение состояния пациентов: нормализует уровни гемоглобина, тромбоцитов; объем печени и селезенки (у неспленэктомизированных больных); купирует костные боли, предотвращает развитие остеонекрозов и костных кризов; приводит к нормализации роста и значительно повышает качество жизни детей с болезнью Гоше.
- имиглюцераза ж,7н ;
- велаглюцераза альфа ж,7н .
Имиглюцераза ж,7н - модифицированная форма кислой ?-глюкозидазы человека, у детей с БГ начальная доза имиглюцеразы составляет:
- при I типе БГ без поражения трубчатых костей скелета - 30 ЕД/кг на 1 введение [6,7,8];
- при I типе БГ с поражением трубчатых костей скелета (костные кризы, патологические переломы, очаги литической деструкции, асептический некроз головок бедренных костей) - 60 ЕД/кг на 1 введение [6,7,8,9,10,11];
- при III типе БГ - 60 ЕД/кг на 1 введение.
Велаглюцераза альфа ж,7н показана для длительного лечения детей с болезнью Гоше I типа. Рекомендуемая доза составляет 30-60 ЕД/кг 1 раз в 2 недели.
Дозу можно корректировать индивидуально, на основании достижения ожидаемого эффекта и его сохранения, однако применение доз выше 60 ЕД/кг не изучено [6,12,13,14,15].
При обследовании сиблингов (братьев и сестер пробанда) могут быть выявлены дети с БГ, не имеющие клинических проявлений. Такие пациенты нуждаются в наблюдении, начинать их лечение необходимо при появлении первых симптомов болезни.
- Не рекомендована ферментная заместительная терапия при II типе БГ, так как не эффективна [1,2,3,5,6].
(Сила рекомендаций - 2; достоверность доказательств - С)
- При развитии проявлений остеопороза для замедления и прекращения потери костной массы, повышения ее прочности, предотвращения переломов костей в комплексной терапии рекомендуется применять: альфакальцидол ж,вк , соли кальция ж,вк [1,3,5,6]
(Сила рекомендаций - 1; достоверность доказательств - С)
Комментарии: в качестве симптоматической терапии скелетных осложнений БГ I типа назначаются анальгетики (во время костных кризов), нестероидные противовоспалительные средства, редко, лишь при наличии показаний, - антибактериальная терапия.
- Не рекомендовано проведение спленэктомии [1,3,5,6].
- При доказанном диагнозе БГ не рекомендованы повторные пункции костного мозга и другие инвазивные диагностические мероприятия (биопсия печени, селезенки) [1,3,5,6].
- Не рекомендовано оперативное лечение костных кризов, которые ошибочно рассматриваются как проявления остеомиелита. При хирургических вмешательствах существует повышенный риск кровотечения и инфицирования [1,2,5,6].
- Противопоказано назначение кортикостероидов с целью купирования цитопенического синдрома [1,2,5,6].
- Не рекомендовано и не обосновано назначение препаратов железа больным с развернутой картиной БГ, так как анемия в этих случаях носит характер «анемии воспаления» [1,2,5,6].
Читайте также: