Синдром Хангарта IV (Hanhart IV) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Синдром Рихнера - Ханхарта развивается на фоне тирозинэмии, связанной с врожденным ферментным дефектом.
Синдром включает в себя ороговевающие бляшки на коленях и локтях, изменение слизистой рта, дебильность и двустороннее поражение роговицы древовидной формы.
При гистологическом исследовании выявлены четкие отличия от истинного древовидного кератита герпетической этиологии.
Наилучший лечебный эффект дают мягкие контактные линзы.
Заболевание встречается редко.
Источник: Каспаров А. А. Офтальмогерпес. М., Медицина, 1994.
РИХНЕРА-ХАНХАРТА СИНДРОМ (син.: тирозинемия П типа, тирозиноз окулокутанный) - наследственный симптомокомплекс, характеризующийся кератодермией, язвенным кератитом и прогрессирующей умственной отсталостью, обусловленными генетическим дефектом фермента тирозинаминотрансферазы.
Недостаточность печеночного фермента тирозинаминотрансферазы приводит к повышению содержания в крови тирозина и отложению в тканях эпидермиса и роговицы его кристаллов, вызывая воспалительную реакцию.
Наследование аутосомно-рецессивное, возможная локализация гена - локус 16q22.1-q22.
Процесс сопровождается нарушением целостности лизосом и выделением протеаз, вызывающих нарушение клеточных структур.
Первые признаки болезни появляются на 1-м году жизни в виде фотофобии, сопровождающейся образованием древовидных изъязвлений роговицы.
Через 1-2 года на коже подошв в местах наибольшего давления (преимущественно на пятках) появляются участки стойкой эритемы с последующим развитием болезненного кератоза, наиболее выраженного в области пяток, свода стопы, заставляющего ребенка ходить на мысочках, щадя область пяток.
Клинически форма кератоза варьирует от плотных массивных роговых масс до пластинчатых чешуйчатых наслоений, иногда с буллезным компонентом; может быть выражен кератоз кончиков пальцев.
Гистологически выявляют гиперкератоз, гранулез, акантоз, эозинофильные включения в шиповатом слое.
Постепенно прогрессирует умственная отсталость.
В моче- гиперсекреция тирозина и его метаболитов.
Диагноз основывается на клинических и лабораторных (повышение тирозина в крови, моче) данных.
Дифференциальный диагноз проводят с различными формами наследственных кератодермий, для которых нехарактерна подобная ферментопатия.
Лечение:
раннее назначение диеты, бедной тирозином и фенилаланином, что позволяет ослабить кожные и глазные симптомы и задержать развитие умственной отсталости. Однако даже в этом случае возможны инвалидизация и летальный исход.
Ханхарта синдром (Hanhart syndrome)
Синонимы: синдром гипоглоссии-гиподактилии, синдром аглоссии-адактилии. Впервые описан в 1950 г. Е. Hanhart.
Минимальные диагностические признаки: микрогения, микроглоссия или аглоссия, редукционные пороки конечностей в виде перомелии.
Клиническая характеристика
Наиболее постоянными признаками являются гипоплазия (или аплазия) языка и гипоплазия нижней челюсти. Встречаются расщелина неба, синехии ротовой полости, сигнатия (сращение челюстей), микростомия, анкилоглоссия (сращение кончика языка со слизистой неба), олигодонтия.
Может наблюдаться одно- или двусторонний паралич черепных нервов. Пороки развития конечностей вариабельны по тяжести проявления и включают синдактилию, олигодактилию, адактилию, частичную или полную ахейрию (аподию), частичную гемимелию предплечья или голени.
Интеллект в некоторых случаях снижен. Популяционная частота неизвестна.
Соотношение полов — M1 :Ж1.
Тип наследования неизвестен, все описанные случаи спорадические.
Дифференциальный диагноз: эктродактилия, ахейроподия, рото-лице-пальцевой синдром, амниотические перетяжки, расщелины неба, латеральных синехий синдром.
«Наследственные синдромы и медико-генетическое консультирование»,
С.И. Козлов, Е.С. Еманова
Читайте далее:
Синдром Ханхарта: фото, симптомы

Синдром Ханхарта — это редкое генетическое заболевание, характеризующееся коротким, не развитым языком (микроглоссия), частичным отсутствием пальцев рук и/или ног (гиподактилия), и чрезвычайно маленькой челюстью (микрогнатия). Дети с этим расстройством часто имеют некоторые, но не все из этих симптомов.
Причина синдрома Ханхарта до конца не выяснена.
Синдром Ханхарта фото
В 1950 году доктор Ханхарт описал трех детей, у которых отсутствовал язык с сопутствующими дефектами конечностей, и было принято название «синдром Ханхарта».
Микроглоссия
Микроглоссия - это недоразвитие или уменьшение размеров языка вследствие врожденных аномалий либо приобретенных заболеваний.
Характеризуется наличием видимого дефекта — язык меньше по размеру.
В легких случаях микроглоссия протекает бессимптомно, в тяжелых отмечается нарушение функций языка, дефекты артикуляции.
Гиподактилия
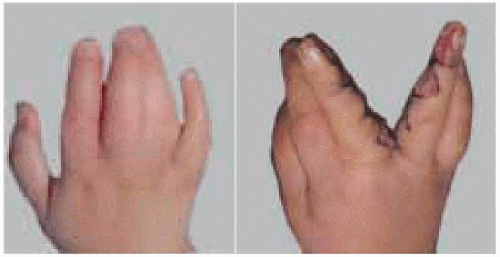
Гиподактилия — аномалия развития: уменьшенное по сравнению с нормой количество пальцев на кисти или стопе.
Микрогнатия
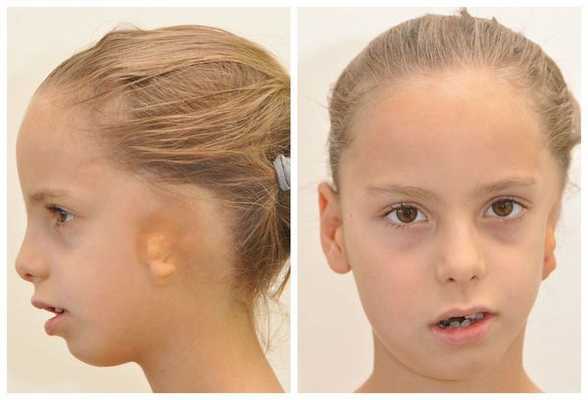
Микрогнатия — это недоразвитие нижней или верхней челюстной кости.
Признаки и симптомы — диагностика заболевания
Черепно-лицевые аномалии у детей с синдромом Ханхарта могут включать в себя:
- маленький рот (микростомия);
- маленькую челюсть или выступающую челюсть (микрогнатия);
- короткий, не полностью развитый язык (гипоглоссия);
- расщелину неба;
- расщелину языка;
- широкий нос;
- увеличенное расстояние между внутренними углами век (телекантус);
- дефекты нижнего века;
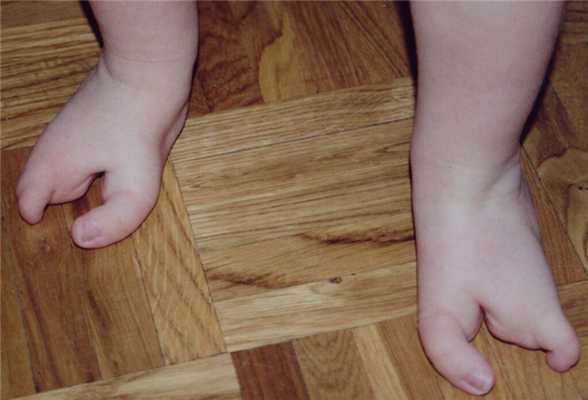
Пораженные дети могут иметь частично отсутствующие или полностью отсутствующие пальцы рук или ног (эктродактилия). Кроме того, части рук и/или ног могут быть деформированы, частично отсутствовать или полностью отсутствовать.
Лечение
Лечение синдрома Ханхарта требует скоординированных усилий команды специалистов. Педиатры, пластические и ортопедические хирурги, стоматологи, логопеды, физиотерапевты должны всесторонне планировать лечение ребенка.
Младенцы с синдромом Ханхарта могут испытывать трудности с приемом пищи в результате пороков развития языка, рта и/или челюсти и паралича черепных нервов, которые необходимо немедленно устранить, чтобы обеспечить правильное питание и рост ребенка.
Аномалии языка, полости рта и челюстной области можно лечить с помощью хирургической коррекции, используя протезы или физиотерапию.
Прогерия
Прогерия - редкое заболевание наследственной природы, характеризующееся преждевременным и ускоренным старением человека. Термин «прогерия» происходит от греческого progeros - «преждевременно состарившийся».
Различают нормальное физиологическое и преждевременное патологическое старение. Физиологическое старение характеризуется постепенным развитием старческих изменений, сокращающим адаптационные резервы организма. Преждевременное патологическое старение проявляется ускоренным темпом данного процесса. При этом биологический возраст человека опережает его паспортный возраст. Ускоренное старение приводит к раннему проявлению возрастной патологии - сердечно-сосудистых и нейро-дегенеративных заболеваний, сахарного диабета и прочих патологий, неблагоприятно влияющих на качество и продолжительность жизни человека.
Одним из первых исследователей, начавших научное изучение проблемы старения, был русский ученый И.И.Мечников. Он полагал, что старение связано с хроническим отравлением организма продуктами гниения, образующихся в кишечнике - ароматическими аминами. В экспериментах И.И.Мечников наблюдал ускорение процесса старения животных, которым вводили индол и крезол - продукты, которые образуются при распаде белков. Поэтому для замедления старения он рекомендовал ограничивать потребление мяса, питаться молочными продуктами, а в качестве антагониста гнилостной бактериальной флоры использовать лактобациллин и проводить стерилизацию пищи.
В 1940 г. А.А.Богомолец выдвинул теорию «возрастной коллоидоклации», согласно которой старение начинается с нарушения функции соединительной ткани в результате необратимых изменений тканевых коллоидов и лабильности белковых молекул.
Позже Гансом Селье была выдвинута гипотеза, согласно которой причиной старения и смерти является исчерпание запаса адаптационной энергии, расходуемой гипоталамо-гипофизарно-надпочечниковой системой при стрессах.
Результаты последних исследований показывают роль апоптоза (программированной клеточной гибели) в процессах старения. В настоящее время определены факторы реализации апоптоза - белок р53, гены - МДМ2, НДМ2, BCL2 и другие
За последние годы было выдвинуто десятки и других гипотез причин старения. Одни гипотезы были основаны на теориях, согласно которым старение - это результат накопления многочисленных повреждающих факторов (соматические мутации, накопление повреждений ДНК, нарушение репарации ДНК, действие свободных радикалов, нарушение иммунных защитных механизмов), другие теории основывались на том, что старение является генетически запрограммированным процессом.
Примерно 7% моногенных наследственных заболеваний имеют признаки ускоренного старения. Среди них выделяет две группы: сегментарные прогероидные синдромы, при которых признаки ускоренного старения выявляются во многих тканях и органах (синдром Хатчинсона-Гилфорда, синдром Вернера и др.), и унимодальные - когда поражается преимущественно один орган или ткань. Примерами унимодальных прогероидных синдромов могут служить семейная гиперхолестеринемия, которая проявляется ранним развитием атеросклеротических повреждений сосудов, а также моногенные доминантные формы болезни Альцгеймера.
Прогероидные синдромы или синдромы преждевременного старения, представляют собой клинически и генетически гетерогенную группу редких наследственных заболеваний, характеризующихся ускоренным старением организма. К прогериям и сегментарным прогероидным синдромам относят свыше десятка заболеваний, однако наиболее ярко признаки преждевременного старения проявляются при синдроме Хатчинсона-Гилфорда (прогерия детского возраста) и синдроме Вернера (прогерия взрослых).
Преждевременное старение в детском возрасте было описано в 1886 г. английским врачом Д. Хатчинсоном у ребенка 6 лет. Оно проявлялось атрофией кожи и её придатков. Термин «прогерия» был введен доктором Н. Гилфордом в 1904 году, давшим клинико-морфологическое описание болезни. Это заболевание встречается очень редко - за всю историю медицины в мире описано всего около 100 случаев.
Симптомы
Детская прогерия начинается с резкого замедления физического развития. Кожа у таких детей сухая и морщинистая, напоминает пергамент. Волосы преждевременно редеют, седеют, брови и ресницы могут отсутствовать. Ногти тонкие и ломкие. Голова значительно увеличивается, лицо становится маскообразным, нос крючковатой формы, уши оттопыренные, определяется экзофтальм. Зубы прорезываются поздно или вообще не появляются. Мускулатура развита очень слабо, подкожно-жировая клетчатка истончена, конечности тонкие, вес таких больных не превышает 20 кг. Половые органы недоразвиты, вторичные половые признаки отсутствуют. Серьезные изменения обнаруживаются в сердечно-сосудистой системе. Уже с 5 лет начинается развитие обширного атеросклероза, развивается тромбоз артерий, в том числе - коронарных с развитием инфаркта миокарда. Отмечаются патологические изменения скелета - дисплазия различных суставов, контрактуры локтевых и коленных суставов.
Детская прогерия сопровождается липодистрофией, генерализованной остеодисплазией с остеолизом и патологическими переломами костей. При этом в головном мозге, печени, почках, эндокринных органах накапливаются отложения жироподобного вещества, развивается склероз. Сначала дети не уступают в умственном развитии своим сверстникам. Но на поздней стадии заболевания - вследствие прогрессирующего атеросклероза - интеллект снижается, происходят неврологические нарушения.
Прогноз при данном заболевании неблагоприятный. Больные редко доживают до совершеннолетия и погибают от сердечно-сосудистых патологий, обычно - инфаркта миокарда, злокачественных новообразований и присоединяющихся инфекционных заболеваний.
Формы
Наиболее ярко признаки преждевременного старения выявляются у двух форм прогерии: детской прогерии Хатчинсона-Гилфорда и прогерии взрослых - синдроме Вернера.
В 1974 г. J.J. Gilkes описал несколько случаев наследственного заболевания, занимающего промежуточное положение между прогерией детской и взрослых, и назвал его «метагерия».
Описаны также и другие многочисленные генетически обусловленные прогероидные синдромы: Видемана-Раутенштрауха, Блума, рестриктивная дермопатия и другие.
Причины
Изучается большое количество факторов, которые могут ускорить физиологический процесс старения, например, экологические факторы, образ жизни, профессиональные и другие. Однако существуют генетически обусловленные формы преждевременного старения (прогерии), при которых скорость патологических изменений, характерных для старения, увеличена во много раз.
Причиной детской прогерии считают врожденную патологию с аутосомно-доминантным типом наследования, связанную с мутацией в гене LMNA. Для развития заболевания достаточно мутированного гена, унаследованного хотя бы у одного из родителей. Этот ген был известен давно, но подробно механизм появления детской прогерии был описан только в 2018 году. Мутация происходит в гене, расположенном в первой хромосоме. В гене закодирована последовательность аминокислот белка преламина А. Данный белок участвует в поддержании каркаса ядер клеток. В результате мутации в 1824 положении гена LMNA происходит замена цитозина на тимин. Это приводит к тому, что из синтезируемого белка «вырезается» часть, и синтезируется измененный преламин А, который получил название прогерин. Прогерин, в отличие от преламина А, не претерпевает дальнейших превращений, а накапливается в ярах клеток, поэтому клетки теряют способность нормально делиться, ткани перестают нормально расти, и развивается их ускоренное старение. Мутация может возникнуть у спонтанно у одного из родителей и затем передаваться последующим поколениям.
Прогерия взрослых впервые была описана немецким студентом-медиком Отто Вернером в 1904 году, когда он наблюдал семью, сочетающих ряд признаков - низкий рост, раннее поседение волос, катаракту и склеродермию. В 1996 году была выяснена генетическая природа данного заболевания, связанная с мутацией в гене WRN. Данный гене расположен в восьмой хромосоме. В этом гене закодирована структура фермента хеликазы. Хеликазы - класс ферментов участвующих в поддержании стабильности клеточного генома, участвующего в разделении двух цепочек ДНК, а также связей в молекуле РНК. На сегодняшний день описано более 80 различных мутаций гена WRN, большинство из которых (65,28%) являются точечными. Мутации в данном гене приводят к нарушению воспроизведения ДНК. Тип наследования прогерии взрослых - аутосомно-рецессивный. Это значит, что для проявления болезни оба родителя должны быть носителями патологических генов, а для того, чтобы болезнь проявилась нужно, чтобы к ребенок получил 2 мутированных гена - один от отца, другой от матери. Если же ребенок получит только один измененный, то он будет «здоровым носителем» болезни.
Методы диагностики
Диагностика прогерии проводится обычно врачом педиатром (детской прогерии) и врачом-генетиком на основании данных анамнеза, клинического осмотра, применения генетических и инструментальных методов обследования. Диагностика направлена на определение клинической формы заболевания, тяжести состояния и возникающих осложнений.
Из анамнеза можно выяснить - наличие у родственников подобных генетических отклонений.
При объективном обследовании могут определяться признаки раннего старения.
Подтверждением наследственного характера болезни является проведение генетического анализа - определение мутаций в генах WRN и LMNA. В крови у больных с детской прогерией отмечается повышенное содержание холестерина и триглицеридов.
Инструментальные методы диагностики - рентгенография, КТ и МРТ различных органов, ЭКГ, ЭХО-КГ, офтальмологические исследования, обследования дерматологом обычно носят второстепенный характер ввиду характерных признаков этих синдромов и проводятся для оценки возникающих осложнений. На рентгенограмме у 80% больных обнаруживается кальцификация мягких тканей, прежде всего в ахилловом сухожилии.
Дифференциальная диагностика синдромов Вернера и Хатчинсона-Гилфорда проводится с другими формами преждевременного старения - неонатальным прогероидным синдром Видемана- Раутенштрауха, прогероидной ламинопатией, прогероидными синдромами, связанными с нарушениями репарации ДНК (пигментная ксеродерма, синдром Коккейна, трихотиодистрофия) и рядом других синдромов (синдромос Секкеля - «птицеголовые карлики», врожденным дискератозом, синдромом Ханхарта).
Основные используемые лабораторные исследования:
- Биохимический анализ крови, включая липидный профиль).
- Поиск мутаций WRN и LMNA в генах во время пренатальной диагностики (хорион, околоплодная жидкость.
- Выявление носительства мутаций WRN и LMNA в генах у будущих родителей при планировании беременности.
Основные используемые инструментальные исследования:
- УЗИ органов брюшной полости (печень, поджелудочная железы, селезенка, почки).
- МРТ и КТ печени, поджелудочной железы, селезенки, головного мозга
- Осмотр врачом-офтальмологом.
- ЭХО-КГ сердца.
Патогенетического лечения детской прогерии в настоящее время не существует.
Были проведены предварительные исследования препарата «Лонафариб», изначально разработанного для лечения рака, но оказавшегося эффективным в отношении прогерии. У всех больных прогерией наблюдалось улучшение по одному из четырех параметров: происходило повышение массы тела, улучшался слух, состояние костных структур, повышалась эластичность кровеносных сосудов.
Для улучшения качества и продления жизни назначают симптоматическое лечение - антиоксидантные препараты, витамин Е и другие лекарства. Для лечения липодистофии, развивающейся при синдроме Хатчинсона-Гилфорда, назначают подкожное введение метрелептина - синтетического аналога лептина, который нормализует липидный профиль и содержание углеводов.
Начиная с 2009 г., многочисленные исследования на мышах продемонстрировали способность антибиотика рапамицина замедлять проявления естественного процесса старения даже при начале терапии на поздних этапах жизни.
В экспериментах на мышах проведена генная терапия, которая продемонстрировала значительное снижение экспрессии мутантного белка прогерина, улучшение структуры и морфологии ядра, а также замедление старения фибробластов.
Осложнения
К основным осложнениям можно отнести раннее развитие атеросклероза с исходом в инфаркт миокарда и инсульт, высокий риск злокачественных новообразований, сахарный диабет, поражение костной ткани (нарушение фосфорно-кальциевого обмена) и метастатическая кальцификация мягких тканей.
Профилактика
Очень важно выявить наличие «патологического гена» у будущих родителей перед принятием ими решения завести ребенка и при планировании беременности.
Другой способ определить возможность развития болезни - уже в период беременности) - выявление мутаций у плода путем взятия ворсин хориона (часть плаценты забирается через шейку матки или через брюшную стенку) или при проведении амниоцентеза. Последняя методика заключается в исследовании клеток плода, находящихся в околоплодной жидкости, получаемых после прокола зародышевой оболочки.
У пациента с прогерией для профилактики низкотравматичных переломов в комбинации назначают бисфосфонаты, препараты витамина D и кальция.
Советы пациенту
Перед принятием решения о деторождении парам при необходимости нужно пойти медико-генетическое консультироване.
Синдром Хантера
Синдром Хантера - наследственное заболевание обмена веществ с Х-сцепленным рецессивным типом наследования, характеризующееся дефицитом лизосомального фермента идуронат-2-сульфатазы и накоплением мукополисахаридов в тканях. При синдроме Хантера отмечается задержка роста, макроцефалия, деформация костно-суставного аппарата, поражение кожи, сердечно-сосудистой и дыхательной системы, гепатоспленомегалия, нарушение слуха, умственная отсталость. С целью диагностики синдрома Хантера проводится консультация генетика, определение экскреции гликозаминогликанов, рентгенография костей и суставов. Пациентам с синдромом Хантера показана пожизненная ферментозамещающая терапия препаратом элапраза.
Общие сведения
Синдром Хантера (мукополисахаридоз II типа) - редкое генетическое заболевание, сцепленное с Х-хромосомой, при котором вследствие ферментной недостаточности происходит неполное разрушение и накопление кислых мукополисахаридов (гликозаминогликанов) в различных тканях. Частота рождения детей с синдромом Хантера составляет примерно 1:100000-150000 новорожденных. В настоящее время в мире насчитывается не более 2000 больных синдром Хантера; по России официальная статистика отсутствует. Синдром Хантера относится к группе орфанных заболеваний, лечение которых, согласно действующему законодательству, должно осуществляться за счет средств федерального и регионального бюджета.
Причины синдрома Хантера
Развитие синдрома Хантера связано с мутацией гена идуронатсульфатазы (IDS), кодирующего лизосомный фермент идуронат-2-сульфатазу. Ген IDS картирован в локусе Xq28 на длинном плече Х-хромосомы; в настоящее время известно более 150 различных его мутаций (точечные мутации, мелкие и крупные делеции, вставки, перестройки и пр.).
В силу сцепленности наследования с Х-хромосомой, синдромом Хантера, как правило, страдают исключительно мальчики (XY). Гетерозиготные женщины в подавляющем большинстве случаев являются носителями мукополисахаридоза II типа без клинических проявлений, однако описаны несколько случаев синдрома Хантера у девочек, связанных с новой мутацией или инактивацией второй, нормальной Х-хромосомы.
Мутации в гене IDS сопровождаются дефицитом или отсутствием фермента iduronate-2-sulfatase (I2S), неполным расщеплением и накоплением в лизосомах клеток практически всех тканей и органов гликозаминогликанов (ГАГ) - дерматансульфата и гепарансульфата.
Классификация синдрома Хантера
Среднетяжелая форма (мукополисахаридоз типа IIВ) составляет примерно 1/3 всех случаев патологии. Клинические проявления синдрома Хантера обычно возникают у детей в 3-8 (иногда 10-13) лет; интеллект обычно сохранен; продолжительность жизни при благоприятных условиях может достигать 50-60 лет. Пациенты с легкой формой синдрома Хантера могут успешно реализовать себя в профессиональной сфере и иметь здоровое потомство.
Симптомы синдрома Хантера
На момент рождения дети с синдромом Хантера выглядят клинически здоровыми. Основная симптоматика развивается в среднем в 2-4 года. До этого возраста проявления заболевания неспецифичны: у детей могут отмечаться повторные риниты, шумное дыхание, пупочные и паховые грыжи, гидроцеле.
Ранним характерным признаком синдрома Хантера служит постепенное изменение внешности ребенка: черты лица становятся грубыми (гаргоилизм); язык, губы и ноздри - большими; кожа - толстой. Облик больного с синдромом Хантера дополняется низкорослостью, увеличением размеров головы (макроцефалией), короткой шеей, аномалиями зубных рядов (редкими зубами). Дети с синдромом Хантера внешне очень похожи друг на друга и напоминают братьев.
Другие ранние признаки мукополисахаридоза II типа включают хриплый низкий голос, увеличение живота, гепатоспленомегалию. Дети с синдромом Хантера склонны к частой заболеваемости ОРВИ, отитами, ларингитом, трахеитом, пневмониями; нередко у них отмечается обструктивное апноэ сна, хроническая диарея. Следствием отложения гликозаминогликанов и липидов в дерме служит возникновение узелково-папулезных высыпаний на коже плеч, бедер, лопаток. Возможно появление «монгольских пятен» в пояснично-крестцовой области, гипертрихоза.
Уже в дошкольном возрасте у детей с синдромом Хантера появляются признаки поражения опорно-двигательного аппарата, выражающиеся в тугоподвижности суставов, развитии кифосколиоза, деформации кистей по типу «когтистой лапы». Движения становятся затруднительными, походка неуклюжей; к юношескому возрасту больные с синдромом Хантера нередко делаются беспомощными инвалидами, прикованными к постели.
Неврологические нарушения, возникающие при синдроме Хантера, включают синдром гипервозбудимости, судороги, сообщающуюся гидроцефалию, спастическую параплегию, задержку речевого развития, прогрессирующую тугоухость. Со стороны зрительной системы обнаруживается помутнение роговицы, атипичный пигментный ретинит. К наиболее поздним признакам мукополисахаридоза II типа относятся кардиологические нарушения: появление шумов в сердце, приобретенные пороки сердца (чаще митральная недостаточность), кардиомиопатия и др.
У ребенка могут наблюдаться изменения психики: обидчивость, агрессивность. В зависимости от типа синдрома Хантера интеллектуальный дефект может быть небольшим или значительно выраженным. Летальный исход у больных с синдромом Хантера обычно наступает от прогрессирующей сердечной или легочной недостаточности.
Диагностика синдрома Хантера
В практике педиатра синдром Хантера встречается исключительно редко. Тем не менее, мукополисахаридоз II типа может быть заподозрен на основании клинических признаков (внешних изменений, манифестации заболевания на 2-4 году жизни, прогредиентного течения и др.). Для подтверждения диагноза дети нуждаются в консультации генетика, анализе клинико-генеалогических данных, проведении молекулярно-генетических исследований.
Важным биохимическим маркером синдрома Хантера служит повышение экскреции мукополисахаридов (дерматансульфата, гепарансульфата) с мочой, дефицит фермента идуронат-2-сульфатазы. Рентгенологические исследования костей черепа, суставов, трубчатых костей, позвоночника демонстрируют дизостоз, остеоартрит, множественные изменения позвонков.
Для обнаружения морфофункциональных изменений со стороны внутренних органов и ЦНС проводится УЗИ органов брюшной полости, ЭКГ, ЭхоКГ, электроэнцефалография, МРТ головного мозга. Морфологическое исследование тканей, полученных в результате биопсии кожи, печени, миокарда и пр., выявляет однотипные изменения - клетки, заполненные гликолипидами.
В семьях с известным генотипом и высоким риском рождения ребенка с синдромом Хантера может проводиться инвазивная пренатальная диагностика - биопсия ворсин хориона, амниоцентез или кордоцентез с определением активности идуронат-2-сульфатазы в полученном материале.
Дифференциальную диагностику синдрома Хантера следует проводить с другими формами мукополисахаридозов (прежде всего, синдромом Гурлера), а также с другими лизосомными болезнями накопления.
Лечение синдрома Хантера
Кроме патогенетического лечения, детям с синдромом Хантера проводятся регулярные медикаментозные курсы симптоматической и корригирующей терапии гепатопротекторами, витаминами, антиоксидантами, цитопротекторами. В комплексную терапию синдрома Хантера целесообразно включать ЛФК, физиотерапию (электрофорез лидазы на суставы, парафиновые аппликации, магнитотерапию, лазеропунктуру), занятия с дефектологом и логопедом.
Прогноз и профилактика синдрома Хантера
Синдром Хантера типа В имеет более благоприятное течение и прогноз; при своевременном и регулярном лечении продолжительность жизни больных может достигать 50-60 лет. При тяжелых формах мукополисахаридоза II типа пациенты обычно погибают до 20 лет от сердечно-сосудистой недостаточности. На сегодняшний день серьезную проблему для больных с синдромом Хантера представляет своевременное получение жизненно необходимого препарата идурсульфазы из-за его высокой стоимости.
Дети с синдромом Хантера нуждаются в наблюдении различных специалистов: детского генетика, педиатра, детского кардиолога, детского невролога, эпилептолога, детского офтальмолога, детского отоларинголога, детского хирурга, детского ортопеда.
Основным методом профилактики синдрома Хантера является медико-генетическое консультирование супружеских пар, имеющих вероятность рождения больного ребенка, а также проведение дородовой диагностики. Следует знать, что у больных с синдромом Хантера рождаются здоровые сыновья, а дочери выступают облигантыми носителями мутантного гена.
Читайте также:
- Влияние половых гормонов на кислородное отравление. Влияние метаболизма на интоксикацию кислородом
- Синдром Белла (Bell)
- Генетика синдрома удлиненного интервала QT. Лечение синдрома удлиненного интервала QT
- Примеры лучевой терапии гемангиомы хориоидеи с четкими контурами
- Введение к описанию буллезных дерматозов