Синдром Курца (Kurz) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 08.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синдром Кушинга: причины появления, симптомы, диагностика и способы лечения.
Синдром Иценко-Кушинга - это сочетание клинических симптомов, вызванных хроническим повышением уровня кортизола или родственных ему кортикостероидов в крови. Болезнь Иценко-Кушинга - это синдром Кушинга, причиной которого служит избыточная гипофизарная продукция адренокортикотропного гормона (АКТГ), как правило, обусловленная небольшой доброкачественной опухолью гипофиза - аденомой. Иногда АКТГ производится опухолью, которая не связана с гипофизом, она может находиться где угодно, чаще - в легких и грудной клетке. Порой злокачественные опухоли хорошо маскируются под железы и начинают вырабатывать гормоны альдостерон и кортизол, что, в свою очередь, приводит к повышению их концентраций в человеческом организме. При этом собственные железы понемногу начинают атрофироваться - таким образом организм старается бороться с избытком гормонов.
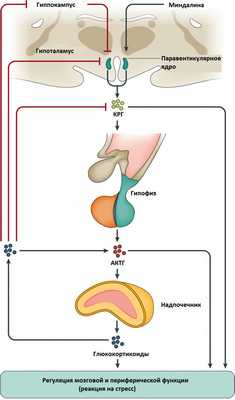
Синдром Иценко-Кушинга также возникает из-за перепроизводства кортизола надпочечниками или употребления больших доз глюкокортикоидных препаратов таких как преднизолон или дексаметазон при лечении ряда болезней (астмы, ревматоидного артрита и некоторых других аутоиммунных патологических состояний). Заболевание может возникнуть в любом возрасте, но чаще всего в 20-40 лет, оно может быть врожденным или приобретенным. Женщины поражаются в 10 раз чаще, чем мужчины.
У пациентов, страдающих алкоголизмом или тяжелыми депрессивными расстройствами, а также во время беременности, иногда наблюдается небольшое повышение уровня гормонов надпочечников и развивается псевдо-синдром Иценко-Кушинга.
Классификация заболевания. Кодирование по МКБ-10
Синдром Иценко-Кушинга (E24):
E24.0. Болезнь Иценко-Кушинга гипофизарного происхождения (гиперсекреция АКТГ гипофизом, гиперадренокортицизм гипофизарного происхождения);
E24.1. Синдром Нельсона;
E24.3. Эктопический АКТГ-синдром;
E24.4. Кушингоидный синдром, вызванный алкоголем;
E24.8. Другие состояния, характеризующиеся кушингоидным синдромом;
E24.9. Синдром Иценко-Кушинга неуточненный.
Симптомы синдрома Иценко-Кушинга
У большинства больных с различными формами гиперкортицизма: АКТГ-зависимыми (болезнь Иценко-Кушинга, аденома гипофиза, АКТГ эктопический синдром) и АКТГ-независимыми формами (аденома коры надпочечника и/или двусторонняя микро-, макроузелковая гиперплазия) клинические проявления заболевания постоянны и зависят от скорости секреции кортизола надпочечниками.
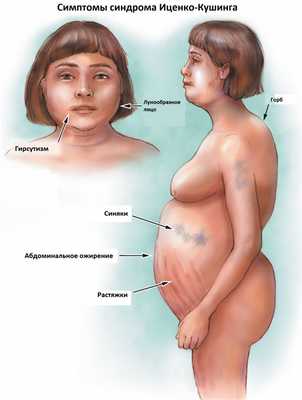
К классическим признакам синдрома Иценко-Кушинга у взрослых относятся «лунообразное» лицо багрово-красного цвета, часто возникают многочисленные угревидные высыпания, центральное ожирение с одновременной потерей жировой ткани на бедрах, ягодицах и руках, истончение кожи и ломкость капилляров, приводящие к легкому и часто спонтанному образованию синяков. За счет неправильного и неравномерного жироотложения происходит необратимая деформация позвоночника, больные сутулятся, происходит нарушение осанки (кифоз, сколиоз). На бедрах, предплечьях, животе можно увидеть растяжки ярко-красного или даже фиолетового цвета, надключичные жировые подушечки и периферические отеки. Часто происходит разрушение костной ткани, отмечается склонность к переломам. У женщин по причине избытка половых гормонов возникают признаки излишнего оволосения по мужскому типу, появляются существенные перебои менструального цикла. У детей самым ранним признаком служит избыточная масса тела при задержке роста.
За счет повышенного уровня кортизола могут возникать гипертония, аритмия, поражение сердца и сосудов, высокий уровень глюкозы в крови, снижение зрения, приступы агрессивности, депрессия, нарушения терморегуляции (именно такие больные очень часто потеют, а также могут мерзнуть в жаркую погоду).
Диагностика синдрома Иценко-Кушинга
Ярко выраженный синдром Иценко-Кушинга не представляет особых диагностических трудностей. Достаточно лишь оценить внешний облик человека и провести с ним беседу. Но заболевание с умеренными проявлениями может вызвать у врача ряд проблем. Всегда следует исключать предварительный прием глюкокортикостероидов пациентом (экзогенный синдром Кушинга). Диагноз ставится клинически, а подтверждается данными лабораторных и визуализирующих методов исследований для установления стадии болезни и выяснения первопричины патологии.
Подтверждение избытка кортизола выполняется строго по показаниям врача одним из четырех методов:
- оценка количества кортизола - определение свободного кортизола мочи в двукратных суточных пробах;
Синонимы: Анализ мочи на кортизол; Анализ суточной мочи на свободный кортизол; Кортизол мочи. Hydrocortisone; Urine cortisol; Free Cortisol Urine Test; Urine Cortisaol Test. Краткое описание теста .
Синдром Куррарино-Сильвермана. Причины, симптомы, диагностика и лечение
Любая структура и система человеческого организма под действием тех или иных неблагоприятных факторов может развиваться неправильно, аномально. К этому предрасположена даже такая фундаментальная, панцирная часть опорно-двигательного аппарата, как грудная клетка. В качестве самостоятельных синдромов в вертебрологии выделено и описано множество вариантов аномального развития грудной клетки. Одни из них встречаются достаточно часто, другие исключительно редки. В среднем, аномальное строение грудной клетки отмечается примерно у 2% людей.
Наиболее распространенные варианты такой аномалии - вогнутая (воронкообразная) и килевидная (выдающаяся кпереди) грудь. Синдром Куррарино-Сильвермана на сегодняшний день является наиболее редкой, по частоте встречаемости, формой врожденной деформации груди. Данный синдром называют также «бычим рогом» и «верхним килем»: грудная клетка выпячена вперед более или менее острым выступом. Иногда выпячивание кпереди сочетается с воронкообразным вдавлением груди вокруг «киля». Названа данная аномалия в честь ученых, которые первыми дали ее подробное клиническое описание и к 60-м годам ХХ века ввели в медицинскую нозологическую лексику.
2. Причины
Основной причиной развития синдрома Куррарино-Сильвермана считают преждевременное окостенение грудины с гипертрофией 2-4 хрящей и/или нескольких ребер. Причины такого деформирующего разрастания, в свою очередь, до сих пор не прояснены. Установлено, что определенную роль играет наследственность, однако этот фактор удается выявить лишь в 25% случаев. Известно также, что у мальчиков синдром Куррарино-Сильвермана встречается вчетверо чаще, чем у девочек. Исследования продолжаются, однако набор материала и статистический анализ проблематичны из-за редкости заболевания.
3. Симптоматика, диагностика
Выпячивание грудной клетки по типу верхнего киля может быть симметричным и асимметричным, односторонним и двусторонним. Аномалия развития грудной клетки проявляется в детстве и окончательно формируется к пубертатному возрасту. Однако если у маленьких детей такая деформация, как правило, не создает какой либо угрозы внутренним органам, то у более старших пациентов нередко наблюдается сдавление сердца и органов дыхания, одышка, утомляемость из-за постоянного дефицита оксигенации, учащенное дыхание, астма и пр. Кроме того, синдром Куррарино-Сильвермана нередко сопровождается врожденными пороками сердца (например, пролапс митрального клапана). Наконец, нельзя не учитывать влияние отчетливого и достаточно выраженного, в некоторых случаях, эстетического дефекта на развивающуюся психику, что может стать источником серьезных психологических нарушений.
4. Лечение
В разные периоды синдром Куррарино-Сильвермана лечили различными способами. Довольно долго в вертебрологии было распространено мнение, что данную деформацию следует корригировать механически - практиковались специальные сдавливающие корсеты, которые пациент должен был носить постоянно в течение двух лет. Однако к настоящему времени представления о целесообразности такого подхода по ряду причин пересмотрены: постоянное ношение жесткого корсета может быть весьма болезненным и, вместе с тем, неэффективным (несмотря на столь продолжительное и сложное для пациента лечение), а то и вредным для прочих органов. На сегодняшний день методом выбора является хирургическая коррекция. Практикуются различные методики торакопластики - по Равичу, по Кондрашину и пр. Некоторые варианты вмешательства, - напр., металлостернохондропластика Тимощенко, - предполагают имплантацию в грудную клетку специальной корригирующей металлической пластины на срок до полугода, однако такая методика также является дискутабельной.
Оптимальным возрастом операции по устранению верхнего киля считают 14-15 лет. В тех случаях, когда синдром Куррарино-Сильвермана сочетается с врожденными пороками сердца или иными сопутствующими аномалиями, чаще всего целесообразна и показана комбинированная операция: устраняется не только вертебрологическая, но и кардиологическая патология.
Курц Рон. Книги онлайн
Рон Курц (Ron Kurtz, 24.02.1934 - 4.01.2011) - автор метода Хакоми телесно-ориентированной психотерапии (который он сейчас называет "основанной на опыте психологией Хакоми), и выдающийся авторитет в передовой психотерапии.
Автор и соавтор трех авторитетных книг («Телесно-ориентированная психотерапия», «Рассказывает тело» и «Раскрывающаяся благодать»), Рон провел сотни тренингов и семинаров по всему миру. Сейчас он ведет четыре трехлетних тренинга «Опорная высота» для профессиональных психотерапевтов и проводит семинары в США, Канаде, Европе, Японии, Новой Зеландии и Австралии.
Работа всей жизни Рона Курца бросила вызов господствующим представлениям о психотерапии и подчеркнула созвучность современной науки о живых системах (которые сейчас обычно называют сложными адаптивными системами) учениям даосизма и буддизма. После докторской диссертации, посвященной математическим моделям обучения и восприятия, он преподавал в госуниверситете Сан-Франциско. Он углубился в основанные на опыте подходы к обучению, тренировке чувствительности, и альтернативные психотерапевтические подходы, в том числе Гештальт, Биоэнергетику, и школы Вильгельма Райха, Моше Фельденкрайза и Иды Рольф.
Основав в 1979 году Институт Хакоми, Рон двенадцать лет проработал его директором. Затем он работал в роли консультанта и создавал новые учебные материалы для обширного персонала Института, который преподает метод Хакоми тысячам студентов по всему миру. Он проработал два года с ведущими разработчиками и руководителями в Кремниевой долине, помогая им освобождаться от ограничивающих личных шаблонов поведения.
В завершающие годы жизни Рон разработал новые структуры процесса обучения, основанные на той идее, что можно создавать сплоченные группы людей с самыми разными профессиями и проблемами. Конечная цель таких групп одна: обеспечить долгосрочную эмоциональную поддержку обычных людей обычным людям. Исследования продемонстрировали, что такие группы увеличивают продолжительность жизни при серьезных заболеваниях. Вторая цель в том, чтобы поощрить социальную работу как неотъемлемую часть личностного и духовного роста.
Книги (3)
С помощью основанных на опыте упражнений, разработанных Роном Курцем для тренингов по основанной на опыте психотерапии Хакоми.
Соавтор: Донна Мартин
Вес упражнения проводятся в парах или в небольших группах из трёх-четырёх человек. Во многих упражнениях только один человек исследует, остальные ассистируют, то есть подают какой-то входной сигнал, например, перемещают руку человека или задают ему вопрос, или физически поддерживая человека во время эмоционального процесса.
Цель здесь в том, чтобы предложить недорогую, самоорганизующуюся систему, которая дополняет и совершенствует обычный процесс «один терапевт - один клиент». Ей могут самостоятельно заниматься как заинтересованные любители, так и профессионалы, проводящие вместе с тем терапию. В систему входят основанные на опыте упражнения, взятые из семинаров и тренингов Хакоми. Они предоставляют выполняющему их человеку возможность исследовать организацию его собственного ума мягким и действенным способом в удобном ему темпе.
Любящее присутствие - это состояние бытия. Это приятно, полезно для здоровья, полезно само по себе. Это состояние добросердечности и добронамеренности. В своих чистейших формах оно духовно и чувствительно к тонким энергиям. Это также наилучшее состояние для того, чтобы оказывать эмоциональную поддержку другому человеку.
Под «эмоциональной поддержкой» мы имеем в виду поддержку тех процессов, которые создают и поддерживают здоровую, счастливую эмоциональную жизнь. Только оглянитесь вокруг, и вы поймете, что это крайне необходимо.
Рон Курц, создатель Хакоми - терапии - один из выдающихся представителей телесно-ориентированной психотерапии.
Метод Хакоми во многом созвучен буддизму и даосизму, с их мягкостью, состраданием, осознанностью и следованием естественной природе вещей. Определенное влияние на формирование метода оказала общая теория систем, которая привнесла в него идею уважения к мудрости каждой личности как живой самоорганизующейся системы.
Метод Хакоми многое почерпнул из современных направлений психотерапии, таких как система Райха, биоэнергетика, гештальт-терапия, психоморная терапия, метод Фельденкрайза, эриксоновский гипноз и нейро-лингвистическое программирование. Хакоми - это синтез философий, техник и подходов, отмеченный уникальным артистизмом, органичной и ясной формой.
Синдром Криглера-Найяра
Синдром Криглера-Найяра - генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина - конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Общие сведения
Синдром Криглера-Найяра - тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами - Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены - большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале - пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Причины
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации - к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление.
Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера - синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Классификация и симптомы синдрома Криглера-Найяра
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением - первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения - нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе - участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой - промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком - он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии. Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Лечение синдрома Криглера-Найяра
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора - в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови - все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия - облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л - при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Прогноз и профилактика
Прогноз синдрома Криглера-Найяра 1-го типа исключительно плохой - из-за полного отсутствия активности фермента уридиндифосфатглюкуронидазы 1 больные умирают на протяжении первого года жизни из-за осложнений ядерной энцефалопатии. Течение СКН-2 зависит от таких факторов, как выраженность проявлений, своевременность диагностики и начала лечения, соблюдения рекомендаций специалистов, наличия или отсутствия сопутствующих заболеваний. В большинстве случаев прогноз относительно благоприятный - больные синдромом Криглера-Найяра 2-го типа могут прожить до преклонного возраста, из характерных проявлений патологии их может беспокоить только желтуха. Профилактика этого состояния возможна только в рамках консультации генетика для родителей, имеющих отягощенную наследственность по этому заболеванию, а также при помощи пренатальной диагностики.
Синдром Кушинга - симптомы, причины, лечение

Синдром Кушинга представляет собой набор симптомов, вызванных высокой концентрацией кортизола, гормона стресса. Синдром Кушинга может появиться у людей всех возрастов. К сожалению, известных методов, способных предотвратить возникновение этого недуга, не существует.
Неизлечимый синдром Кушинга и болезнь Кушинга вызывают другие заболевания, такие как гипертония, диабет и остеопороз, и увеличивают риск смерти. Поэтому, чтобы понимать, находитесь ли вы в группе риска, важно знать, что вызывает синдром Кушинга. Правильно подобранное лечение дает шанс на нормальную жизнь.
Что такое синдром Кушинга
Синдром Кушинга или гиперкортицизм - это совокупность симптомов заболевания, возникающих из-за избытка кортизола в организме. Название патологии произошло от имени выдающегося нейрохирурга - Харви Кушинга, который первым наблюдал и описывал это заболевание.
Наиболее частая причина синдрома Кушинга — прием глюкокортикостероидов - препаратов от других заболеваний, повышающих уровень гормона стресса в крови.
Выделяют два варианта синдрома Кушинга:
- ActH зависимый — болезнь Кушинга. В этом случае аденома гипофиза (железа, расположенная в средней черепной ямке) выделяет чрезмерное количество АКТГ — адренокортикотропного гормона (кортикотропина). Этот гормон оказывает косвенное влияние на белковый, углеводный и минеральный баланс организма. Однако его слишком высокая концентрация оказывает негативное влияние на здоровье.
- Независимый от ACTH . Патология обусловлена возникновением опухолей коры надпочечников и экстравазальных опухолей (аденома или рак).
Кто чаще всего заболевает синдромом Кушинга?
Синдром Кушинга чаще всего встречается у людей в возрасте от 25 до 50 лет. В основном это касается женщин. Однако синдром Кушинга может возникать и у детей, особенно если они принимают глюкокортикоиды. У малышей обычно отмечаются те же симптомы, что и у взрослых. Дополнительный симптом, характерный для детей, — карликовость.
Симптомы синдрома Кушинга
Симптомы синдрома Кушинга хорошо заметны, однако, они не являются симптомами, характерными только для этого заболевания, из-за чего оно не всегда сразу диагностируется.
Люди, страдающие синдромом Кушинга, чаще всего сталкиваются с такими проблемами, как:
- увеличение веса и увеличение жира в организме, особенно в области шеи и лица (так называемая «шея буйвола» и лицо, похожее на луну);
- характерный силуэт - толстое лицо, тучный торс и очень тонкие конечности (руки и ноги);
- покраснения кожи лица;
- сильно видны растяжки на животе, бедрах, ягодицах и груди;
- проблемы с кожей - акне;
- нарушения менструального цикла и чрезмерные волосы у женщин;
- депрессия, нарушения сна и эмоциональная нестабильность;
- артериальная гипертензия;
- сахарный диабет;
- ломкость костей и остеопороз;
- частые и рецидивирующие инфекции;
- подверженность появлению синяков на теле;
- задержка роста костей у детей, то есть карликовость.
Большинство из этих клинических признаков проявляются в запущенной фазе заболевания. Поначалу могут возникать только некоторые из них, что затрудняет диагностику синдрома Кушинга.
Причины синдрома Кушинга
Наиболее частая причина этого заболевания — применение глюкокортикоидных препаратов, увеличивающих количество стероидных гормонов в организме. Эти лекарства принимают во время лечения астмы, ревматоидного артрита, у пациентов после трансплантации органов (они предотвращают отторжение пересаженного органа).
Гормон стресса (кортизол), вырабатывающийся в коре надпочечников, способствует формированию синдрома Кушинга. Если надпочечники, контролируемые гипофизом, вырабатывают его в слишком больших количествах, причиной чего могут быть опухоли гипофиза или надпочечников, то чрезмерная концентрация кортизола действует неблагоприятно.
Синдром Кушинга также может вызвать болезнь Кушинга, являющуюся одним из видов гиперактивности передней доли гипофиза. Тогда причина чрезмерной концентрации АКТГ — опухоль.
Как диагностировать синдром Кушинга?
Для подтверждения возникновения синдрома Кушинга следует провести ряд лабораторных анализов. К какому врачу следует обратиться, чтобы диагностировать синдром Кушинга? При подозрении на патологию лучше всего обратиться к эндокринологу. Женщины могут обратиться к гинекологу, так как эти специализации тесно связаны.
Врач, на основании собеседования, назначит скрининговые тесты:
- Определение уровня кортизола в крови - уровень кортизола у здоровых людей вечером низкий, в то время как у людей с синдромом Кушинга его концентрация ночью повышается. При таком обследовании необходимо проводить анализ вечером.
- Тест на ингибирование дексаметазона. Анализ заключается в приеме 1 мг дексаметазона перед сном. На следующее утро нужно сделать анализ крови натощак, чтобы определить уровень кортизола. Обследование не требует пребывания в стационаре и может проводиться амбулаторно.
- Ежедневное выведение свободного кортизола с мочой. Для выполнения этого анализа, после первой мочи, сданной утром, соберите мочу в течение 24 часов в емкость. Последняя моча для сбора - это поступление с первого утреннего мочеиспускания (сразу после вставания) на следующий день. Рекомендуется определять свободный кортизол не менее чем в двух ежедневных сборах мочи.
- Тест стимуляции CRH . Кортикотропин-рилизинг-гормон принимается в виде инъекции. Тест состоит из трех определений концентраций кортизола и АКТГ. Их концентрацию измеряют впервые до введения CRH, затем через 30 минут после инъекции и через 60 минут после. У здоровых людей максимальная концентрация АКТГ наступает через 30 минут, а кортизола - через 60. Пациенты с синдромом Кушинга чаще всего не реагируют на CRH.
Если результаты показывают, что в организме наблюдается повышенная концентрация кортизола, то необходимо провести несколько визуальных тестов, подобранных в зависимости от возможной причины синдрома Кушинга. К ним относятся:
- магнитно-резонансная томография гипофиза (при подозрении на болезнь Кушинга);
- компьютерная томография или магнитно-резонансная томография надпочечников (если предыдущие результаты анализов указывают на наличие опухоли надпочечников);
- компьютерная томография грудной клетки и живота;
- УЗИ щитовидной железы;
- радиоизотопные тесты.
Три последних исследования визуализации предназначены для того, чтобы помочь найти опухоль, секретирующую избыток адренокортикотропного гормона (АКТГ).
Синдром Кушинга и болезнь Кушинга - чем они отличаются?
Болезнь Кушинга отличается от синдрома Кушинга, поэтому эти понятия невзаимозаменяемые. Хотя симптомы синдрома Кушинга такие же, как и в случае Болезни Кушинга и диагноз схож, причины их формирования различны. Болезнь Кушинга вызывается опухолью гипофиза, расположенной в средней черепной яме. Заболевание лечится хирургическим путем.
В диагностике этих двух недугов может помочь тест на ингибирование дексаметазоном с использованием увеличенной дозы - 8 мг дексаметазона. Болезнь Кушинга может вызвать синдром Кушинга.
Синдром Кушинга - лечение
Метод лечения синдрома Кушинга зависит от его причины. Если синдром является следствием болезни Кушинга, которая вызвана аденомой, то необходима операция. Через сфеноидальный синус опухоль уничтожается лазером или ионизирующими лучами.
После такой процедуры нужно находиться под постоянным наблюдением эндокринолога, так как есть риск рецидива заболевания. Может случиться так, что операция не удалась или удаление аденомы невозможно. Затем у пациентов применяется лучевая терапия.
Синдром Кушинга, вызванный экструзионными опухолями, лечится хирургическим путем или с применением лучевой терапии или химиотерапии.
От опухоли надпочечников, которая также может быть причиной синдрома Кушинга, можно избавиться, удалив пораженную железу. Обычно после такой операции нужно принимать лекарства, которые будут регулировать уровень кортизола, ведь оставшаяся одна железа может поначалу выделять слишком мало гормона.
Если причина недуга — глюкокортикоидные препараты, то следует обратиться к врачу, который снизит их дозу. Запрещено прекращать прием лекарств самостоятельно, так как это может стать причиной развития других заболеваний.
Читайте также:
- Анатомия: Нижняя носовая раковина, носовая кость, слезная кость, сошник, скуловая кость
- Способы исследования психотерапевтических методов
- Семенной канатик. Элементы семенного канатика. Круглая связка матки. Оболочки круглой связки матки.
- Диагностика изоиммунизации беременной. Тактика ведения беременности при изоиммунизации.
- Руминация и извращение вкуса у детей. Причины