Синдром миопатический - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 01.02.2026
Миотонические расстройства объединяют наследственные нервно-мышечные заболевания, в основе которых лежит миотонический тип нарушения движений или миотонический феномен. Миотонический феномен характеризуется своеобразным состоянием мускулатуры, при котором после активного напряжения мышц возникает тонический спазм с затруднением расслабления - сократившаяся мышца как бы стремится удержать свое состояние напряжения, фаза расслабления растягивается на 5-30 секунд. Наибольшие затруднения больные испытывают при первых движениях, повторные движения совершаются свободнее и через некоторое время могут нормализоваться. Миотонии - это группа заболеваний, различающихся как по клинической картине, так и по типу наследования. Характерным для миотонии является гипертрофия мышечных волокон, чрезмерное ветвление нервных окончаний.
Протокол "Миотонические расстройства"
Код по МКБ-10: G71.1
Дистрофия миотоническая (Штейнера)
- Доминантное наследование (Томсена)
- Рециссивное наследование (Беккера)
При необходимости идентифицировать лекарственное средство, вызвавшее поражение

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
Классификация D. Gardner - Mеtdwin, Y. Walton:
- дистрофическая миотония (миотоническая дистрофия);
- врожденная миотония (аутосомно-доминантная форма Томсена);
- врожденная миотония (аутосомно-рециссивная форма);
- хондродистофическая миотония, синдром Шварца-Джампела;
- врожденная парамиотония Эйленбурга;
- парамиотония с параличом, с параличом возникающим на холоде.
Значительно расширили и систематизировали классификацию миотонических заболеваний В.С. Лобзин с соавторами:
1. Наследственные формы миотонии.
А. Стационарные медленно прогрессирующие формы:
- врожденная миотония Томсена (аутосомно-доминантный тип наследования);
- врожденная миотония Томассена-Беккера (аутосомно-рециссивный тип наследования);
- приобретенная миотония Тальма (спорадические случаи);
- атрофическая (дистрофическая) миотония или миотоническая дистрофия Гофмана-Россолимо-Штейнерта-Куршманна (аутосомно-доминантный тип наследования);
- миотоническая дистрофия Беккера (аутосомно-рециссивный тип);
- клинические варианты миотонической дистрофии (атрофическая миотония «без атрофии», миотония лица и шеи Эрбсле, момсимптомный вариант - « миотоническая катаракта» неонатальная форма дистрофической миотонии).
Б. Периодические (рецивидирующие) формы миотонии:
- интермиттирующая миотония Марциуса-Ганземана (аутосомно-доминантное наследование);
- врожденная парамиотония с холодовыми параличами Эйленбурга (аутосомно-доминантное наследование);
- врожденная парамиотония без холодовых параличей Де Ионга (аутосомно-доминантное наследование);
- эпизодическая наследственная миотоническая адинамия Беккера (аутосомно-доминантное наследование);
- периодический парамиотонический паралич Беккера (аутосомно-доминантное наследование).
2. Миотонические синдромы:
- миотонические синдромы у больных миопатиями;
- миотонические синдромы при периодическом параличе и эпизодической адинамии Гамстроп;
- миотонические синдромы при органических болезнях ЦНС;
- миотонические синдромы при заболеваниях внутренних органов;
Диагностика
Диагностические критерии
Жалобы и анамнез: нарушение движений, затруднение расслабления мышц, спазмы жевательных, глоточных мышц, усиление спазмов на холоде, при внутренней напряженности, симптом валика, атрофии, контрактуры, затруднение при беге, ходьбе по лестнице. Начало заболеваний может быть в любом возрасте, чаще в детском или юношеском возрасте.
Физикальные обследования
Дистрофия миотоническая (Штейнерта) Наследуется по аутосомно-доминатному типу.
Характеризуется:
- миотоническим типом нарушения движений;
- миопатическим синдромом с характерным распределением амиотрофий (поражение мускулатуры лица, шеи, дистальных отделов рук и ног);
- вовлечение в процесс эндокринной и вегетативной систем. А также снижение зрения вследствие катаракты.
Выделяют 4 формы заболевания: развернутая форма, стертая форма, неонатальная форма, атрофическая миотония без миотонии.
Хондродистрофическая (Швартца-Джампеля). Наследуется по аутосомно-рециссивному типу. Характерно сочетание миотонического синдрома с повышенной механической и электрической возбудимостью и мышечной гипертрофией. Отмечается длительное сокращение различных мышечных групп с болевым синдромом типа крампи. Кроме мышечных феноменов выявляются различные аномалии развития скелета - карликовость, короткая шея, эпифизрная дисплазия, кифоз, сколиоз, недоразвития лицевого скелета.
Лекарственная миотония - приступы мышечной слабости отмечаются у больных принимавших различные медикаменты, способствующие выведению калия из организма: диуретики, слабительные, лакричник. Гипокалиемический эффект лакричника обусловлен с альдостероноподобным метаболическим эффектом препарата. При отравлении барием также развивается пароксизмальная миоплегия. Отличительной особенностью бариевой интоксикации является вовлечение гладкой мускулатуры, повышение артериального давления, сердечная аритмия. Приступы сопровождались гипокалиемией. Механизм развития миоплегии при бариевой интоксикации заключается в способности бария блокировать выход калия из мышечных клеток.
Симптоматическая - наиболее частой причиной вторичной формы миоплегии является тиреотоксикоз. Приступы тиреотоксической миоплегии сопровождались снижением сывороточного калия, во внеприступном периоде концентрация калия была в норме, характерным для тиреотоксической миоплегии являются приступы в вечерние часы.
При первичном гиперальдостеронизме дефицит калия, обусловленный повышенным выделением с мочой, способствует появлению у больных нарастающей мышечной слабости, на фоне которой возникают приступы миоплегии. Пароксизмальная миоплегия как осложнение желудочно-кишечных заболеваний является следствием гипокалиемии, связанной с потерей калия при поносах и рвоте. При заболеваниях почек т.к. нарушение функции почек может сопровождаться как гипокалиемией, так и гиперкалиемией, поэтому возможны приступы паралича как гипер-, так и гипокалиемического происхождения. При гипоталамической патологии механизм развития мышечной слабости связан с гипернатриемией вследствие нарушения освобождения антидиуретического гормона.
Врожденная миотония Томсена - наследуется по аутосомно-доминантному типу. Заболевание может проявиться уже на первом году жизни. Основной симптом миотонии - нарушение движений, заключающееся в том, что после сильного сокращения мышц расслабление резко затруднено, но при повторении движений оно становится все более свободным и, наконец, нормальным. Спазмы усиливаются при охлаждении или внутреннем напряжении. При постукивании по мышцам на месте удара образуется валик. Симптом валика может наблюдаться и в мышцах языка. Трудность расслабления глоточных мышц вызывает нарушение глотания. Мышечная система развита, больные имеют атлетический вид. Прогрессирует крайне медленно, психика не страдает.
Аутосомно-рециссивная форма Беккера или генерализованная миотония, отличается генерализованным поражением мышц, наличием мышечной слабости, атрофией мышц предплечья и шеи, прогрессирующим течением, постепенным вовлечением в патологический процесс групп мышц в восходящем порядке, иногда отмечается усиление миотонии на холоде, гипогонадизм, утолщение костей черепа.
Нейромиотония (синдром Исаакса). Характеризуется значительным повышением мышечного тонуса постоянного типа, захватывающего преимущественно дистальные отделы рук и ног, больше флексорные группы мышц. Процесс может распространяться и на проксимальные отделы конечностей, а также мышцы лица, глотки, но мышцы туловища и шеи вовлекаются очень редко. В результате постоянной мышечной гипертонии, не исчезающей даже во сне, быстро формируются контрактуры, особенно в кистях и стопах. Характерно наличие постоянных крупных фасцикулярных подергиваний в мышцах рук и ног (миокимии). Активные движения затруднены из-за увеличения мышечных спазмов, повторные движения усиливают ригидность. Мышцы очень плотны на ощупь, быстро развивается гипертрофия, хотя в тяжелых контрактурах могут развиться контрактуры. Заболевание начинается в любом возрасте, течение медленно прогрессирующее с постепенной генерализацией процесса.
Парамиотония врожденная. Наследуется по аутосомно-доминантному типу, имеет четко семейный характер. Начало заболевания с первых дней. Характеризуется появлением спазма мышц после общего охлаждения (пребывание на холоде в зимнее или осеннее время, купание в холодной воде) Появляется спазм в мышцах лица - круговой мышце глаз, рта, жевательной мускулатуре, нередко в глазодвигательных мышцах. Как и при миотонии после активного сокращения, возникает резкое затруднение расслабления, при повторных движениях спазм имеет тенденцию к усилению. У большинства больных развиваются парезы и даже параличи в пораженных мышцах длящихся от нескольких минут до нескольких часов и даже суток. Прекращение воздействия холода, интенсивное согревание ускоряют нормализацию функций. Патогенез заболевания неизвестен.
Гиперкалиемическая форма пароксизмальной миоплегии. Первые приступы появляются в первые 5 лет. Первыми симптомами является чувство тяжести в конечностях с акропарастезией в области лица. Затем развивается мышечная слабость, которая достигает степени паралича в течение получаса и заканчивается столь же быстро, но иногда может длиться несколько суток. Приступы возникают чаще в дневное время. Провоцирующий эффект - отдых после физической нагрузки, переохлаждение и голодание.
Нормокалиемическая форма. Приступы миоплегии не сопровождались изменениями содержания сывороточного калия. Приступы различной выраженности, вплоть до тетраплегии, когда больные не могли самостоятельно повернуться в постели, часто при этом имелась слабость жевательной мускулатуры. Полный паралич может длиться около недели, и еще 2-3 недели требовалось для восстановления.
Лабораторные исследования: изменения уровня калия в сыворотке крови -гиперкалиемия или гипокалиемия.
Инструментальные исследования:
1. КТ головного мозга для исключения органического поражения.
2. ЭЭГ - изменения ЭЭГ неспецифичны.
3. ЭМГ - для миотонии различного происхождения характерны так называемые миотонические разряды, представляющую собой высокочастотную активность, состоящую из позитивно-негативных спайков, положительных острых волн и частично потенциалов, соответствующих разрядам ДЕ. Частота активности - 15-150/с. Амплитуда и частота потенциалов совершают характерные флюктуации в виде нарастания и снижения. Миотонические разряды вызываются введением электрода, простукиванием по мышце, электрической стимуляцией нерва и мышцы, произвольным сокращением. При повторных произвольных сокращениях выраженность миотонических разрядов уменьшается.
При миотонии Томсена характерна спонтанная активность в виде «миотонического разряда», свидетельствующего о повышенной возбудимости миотонической мышцы и наклонности давать повторные ответы на любые формы раздражения. При миотонической дистрофии отмечаются характерные для первично-мышечных дистрофий укорочения длительности потенциалов ДЕ, снижение их амплитуды, увеличение процента полифазных колебаний.
4. ЭКГ - выявляет аритмию, нарушение обменных процессов в миокарде.
Показания для консультаций специалистов:
- логопед - для выявления нарушения речи, дизартрий;
- психолог - для определения психического статуса ребенка;
- ортопед - для выявления контрактур, костных деформаций;
- протезист - для подбора ортопедической обуви, фиксирующих лонгет;
- окулист - осмотр глазного дна, выявление катаракты, нарушений зрения;
- кардиолог - выявление обменно-дистрофических изменений в миокарде.
Минимум обследования при направлении в стационар:
1. Общий анализ крови.
2. Общий анализ мочи.
3. Кал на яйца глист.
Основные диагностические мероприятия:
Перечень дополнительных диагностических мероприятий:
1. Компьютерная томография головного мозга.
5. МРТ головного мозга.
7. R-графия грудной клетки.
8. УЗИ органов брюшной полости.
10. ЛОР сурдолог.
Дифференциальный диагноз
Заболевания
Клиника
Чувствительность
ЭМГ
С появления миотонического типа нарушений движений, характерное распределение атрофий и парезов с участием мышц лица и шеи, наличие повышенной механической возбудимости мышц
Характерна спонтанная активность в виде «миотонического разряда», свидетельствующего о повышенной возбудимости миотонической мышцы и наклонности, давать повторные ответы на любые формы раздражения. При миотонической дистрофии отмечаются характерные для первично-мышечных дистрофий укорочение длительности потенциалов ДЕ, снижение их амплитуды, увеличение процента полифазных колебаний
Невральная амиотрфия Шарко-Мари
С появления амиотрофии, мышечной слабости, парестезии, болезненность при пальпации по ходу нервных стволов, вальгусная установка и деформация стоп, походка типа «степпаж»
Расстройство поверхностной чувствительности
Наблюдаются изменения, характерные для хронического денервационного процесса. При произвольном сокращении регистрируется разреженная активность. Наблюдаются спонтанные электромиографические феномены в виде фибрилляций, положительных острых волн и фасцикуляций
Дистальные формы миодистрофий
Слабость, атрофия в мышцах голени и стоп, затем дистальных отделов верхних конечностей, мускулатура лица интактная, развиваются кардиомиопатии
Изменение на ЭМГ указывают на дегенеративное поражение самой мышечной ткани, основным признаком является сохранность скорости проведения возбуждения по нервному стволу
Лечение
Тактика лечения
Цель лечения: улучшение двигательной активности, социальная адаптация, профилактика патологических поз и деформаций.
Немедикаментозное лечение:
1. Диета с ограничением калия (картофель, урюк, чернослив и др.), употребление продуктов с повышенным содержанием кальция, необходимо исключить холодную пищу и другие виды переохлаждения.
3. ЛФК дозированно.
6. Кондуктивная педагогика.
7. Занятия с логопедом, психологом.
Медикаментозное лечение:
1. Нейропротекторы: церебролизин, актовегин, пирацетам, пиритинол, гинкго-билоба, гопантеновая кислота, глицин.
2. Стимулирующая терапия: прозерин, дибазол, галантамин, оксазил.
3. Ангиопротекторы: винпоцетин, циннаризин.
4. Витаминотерапия: витамины группы В - тиамин бромид, пиридоксин гидрохлорид, цианкобаламид, фолиевая кислота, токоферол, ретинол, эргокальциферрол.
5. Седативные препараты: ново-пассит, ноофен.
6. Миорелаксанты: тизанидин, баклофен, толперизон.
7. Препараты кальция: хлористый кальций, глюконат кальция.
8. Стероидные анаболические гормоны: ретоболил, нейробол, метиландростендиол.
9. Противосудорожные препараты: дифенин.
10. Диуретик - ацетазоламид.
Дальнейшее ведение: регулярное занятия ЛФК, обучение родителей навыкам массажа, ношение ортопедической обуви.
Перечень основных медикаментов:
1. Актовегин ампулы по 80 мг 2 мл
2. Пирацетам, ампулы по 5 мл 20%
3. Пиридоксин гидрохлорид ампулы по 1 мл
4. Фолиевая кислота, таблетки 0,001
5. Цианокобаламин, ампулы 200 и 500 мкг
Дополнительные медикаменты:
1. Аевит, капсулы
2. Аспаркам, таблетки
3. Ацетозоламид, таблетки 0,25
4. Винпоцетин, таблетки
5. Гинкго-Билоба, таблетки 40 мг
6. Глицин, таблетки 0,1
7. Глюконат кальция, ампулы 10%, таблетки по 0,5
8. Гопантеновая кислота, таблетки 0,25
9. Дибазол, таблетки 0,02
10. Дифенин, таблетки 0,1
11. Луцетам, таблетки 0,4
12. Магне В6, таблетки
13. Метиландростендиол 0,025
14. Неробол, таблетки 0,005
15. Ново-пассит, таблетки, сироп
16. Ноофен, таблетки 0,25
17. Панангин, таблетки
18. Пирацетам, таблетки 0,2
19. Пиритинол, суспензия или таблетки 0,1
20. Ретаболил, ампулы 5% 1 мл
21. Тиамин хлорид, ампулы, 1 мл 5%
22. Толпиризон, ампулы 1 мл (мидокалм)
23. Толпиризон, таблетки 0,05
24. Хлористый кальций, ампулы 10 мл 10%
25. Церебролизин, ампулы 1 мл
26. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
1. Улучшение двигательной и речевой активности.
2. Уменьшение мышечного спазма.
3. Улучшение глотания и жевания.
4. Пополнение активного и пассивного запаса слов.
5. Улучшение эмоционального и психического тонуса ребенка.
Госпитализация
Показания к госпитализации (плановая): двигательные расстройства из-за мышечного спазма, нарушение расслабления мышц, спотыкающаяся походка, мышечные спазмы в глоточных, жевательных мышцах, атрофия мышц лица, ранняя катаракта с атрофией мышц, скованность, затруднение расслабления мышц, контрактуры.
Митохондриальная миопатия
Митохондриальные миопатии — это группа заболеваний мышц, которые возникают в результате нарушения функции тканевого дыхания при патологиях митохондрий. Болезни проявляются нарастающей мышечной слабостью, атрофией мускулатуры, прогрессирующими двигательными расстройствами, которые могут сопровождаться судорогами, поражениями сердца, ухудшением слуха и зрения. Диагностика предполагает лабораторные (исследование мышечных биоптатов, генетическое тестирование, биохимические анализы), инструментальные методы (ЭМГ, церебральное МР-сканирование). Лечение включает симптоматические препараты, физиотерапию, ЛФК.
МКБ-10

Общие сведения
Термин «митохондриальная миопатия» объединяет в себе несколько патологий со сходным патогенезом. Наиболее часто дети страдают синдромами MELAS, MERRF, синдромом Кернса-Сайра с хронической прогрессирующей наружной офтальмоплегией. Изучение молекулярно-генетических особенностей этих болезней началось в 1980-х гг. Распространенность патологий у детей составляет около 11,5 случая на 100 тыс. населения. Из-за мультисистемности поражения, клинического полиморфизма, вариабельности течения этот вид миопатии представляет серьезные трудности в диагностике.

Причины
Миопатии обусловлены наследственными либо спорадическими мутациями в митохондриальной ДНК или в ядерных генах, контролирующих работу митохондрий. Известно более 300 вариантов генных дефектов — однонуклеотидных замен, делеций, вызывающих нарушения функционирования митохондрий. Митохондриальные миопатии имеют ряд генетических аспектов, выделяющих их среди всех наследственных заболеваний. Принципиальные различия:
- Материнское наследование. Эмбрион получает всю цитоплазму с содержащимися в ней органеллами от матери, поэтому только она может передать ребенку мутантную мтДНК. В то же время, при мутациях ядерной ДНК наследование происходит по аутосомно-рецессивному, аутосомно-доминантному или Х-сцепленному типу.
- Гетероплазмия. При митохондриальной миопатии в клетках мышечной ткани одновременно присутствует мутантный и нормальный генетический материал в разном процентном соотношении, чем объясняется вариабельность клинических проявлений у членов одной семьи при наследственной форме болезни.
- Митотическая сегрегация. При делении клеток, содержащих мутантные митохондриальные гены, мтДНК распределяется между дочерними клетками случайным образом, неравномерно.
Патогенез
Митохондрия — универсальная органелла, присутствующая во всех клетках, кроме эритроцитов. Она имеет дыхательную цепь, которая включает 5 ферментных комплексов из нескольких десятков субъединиц каждый. Ферменты обеспечивают окислительное фосфорилирование для синтеза АТФ — аденозинтрифосфата, выступающего основным источником энергии в организме.
При митохондриальных заболеваниях может быть 4 варианта патогенетических механизмов развития. Как правило, при поражениях мышц наблюдаются дефекты электронного транспорта и окислительного фосфорилирования, что сопровождается нарушениями образования энергетических молекул. Другие варианты расстройств включают нарушения обмена пирувата, дефекты метаболизма жирных кислот, дисфункцию цикла Кребса.
При миопатиях отмечаются полисистемные расстройства. Мышечная и нервная ткань, больше других зависимые от энергопроизводства, страдают в первую очередь. Поражение проявляется нарушениями обмена веществ в мышцах, дегенеративными процессами, атрофией миофибрилл и их замещением соединительной тканью. Со временем патологический процесс распространяется на сердечную мышцу, эндокринную систему, почки и печень, что определяет различные «маски» миопатии.
Симптомы митохондриальной миопатии
Патологии манифестируют у детей раннего возраста, иногда они присутствуют с рождения. Общим признаком является миопатический синдром, который включает прогрессирующую мышечную слабость, снижение тонуса скелетной мускулатуры, непереносимость физических нагрузок. Типично отставание в моторном развитии: дети поздно начинают сидеть, ползать, ходить, у них сохраняется неуклюжесть движений, проблемы с поддержанием равновесия.
Клинические особенности определяются типом заболевания. При синдроме MERRF мышечная слабость сопровождается разнообразными судорожными приступами (атоническими, тонико-клоническими, миоклоническими), расстройствами координации вследствие мозжечковой атаксии. Для синдрома MELAS характерны повторные инсульты, умственная отсталость, нейросенсорная тугоухость.
У детей распространен синдром Кернса-Сайра, при котором клинические признаки дополняются расстройствами глотания, нарушениями работы проводящей системы сердца, снижением слуха. Хроническая прогрессирующая наружная офтальмопатия может возникать как компонент болезни Кернса-Сайра, так и развиваться изолированно, что чаще бывает в старшем возрасте.
Осложнения
Отличительными особенностями миопатического синдрома являются необратимость, неуклонное прогрессирование. Сначала мышечная слабость появляется в проксимальных отделах конечностей, затем поражает все тело ребенка: в процесс вовлекается гладкая мускулатура органов дыхания и пищеварения, что чревато дыхательной недостаточностью, аспирационными пневмониями, тотальным параличом. Такие пациенты теряют способность к самообслуживанию, требуют круглосуточного ухода.
Миопатии осложняются деформациями позвоночника, искривлениями нижних конечностей на фоне слабости мышечного корсета. Вследствие атрофии зрительных нервов у больных с синдромом MERRF возникает слепота. Опасным последствием многих вариантов митохондриальной патологии являются инсульты, эпилептический статус, мозговой отек, которые становятся основными причинами летального исхода.
Первичное обследование детей с подозрением на митохондриальную миопатию проводится у невролога, для уточнения диагноза показана консультация генетика. При осмотре учитывается неврологический статус ребенка, показатели мышечной силы и тонуса, уровень развития когнитивных навыков. Постановка диагноза требует комплексного обследования, включающего следующие методы:
- Электромиография. Исследование демонстрирует уменьшение амплитуды и длительности регистрируемых потенциалов двигательных единиц (ПДЕ), что указывает на разнокалиберность мышечных волокон с их мозаичной атрофией, гипертрофию небольшой части миофибрилл.
- Исследование биоптатов мышц. Патогномоничным признаком митохондриальной миопатии является феномен «рваных красных волокон», который определяется при специальной окраске биоптатов трихромом по Гомори.
- МРТ головного мозга. Нейровизуализация назначается при подозрении на центральный характер мышечных нарушений, для исключения сопутствующих нейродегенеративных поражений ЦНС, которые нередко встречаются при митохондриальных болезнях.
- Цитоморфоденситометрия. Анализ необходим для оценки активности митохондрий в лимфоцитах ребенка, показывает снижение числа органелл при увеличении их объема, уменьшение оптической плотности гранул, нарушения ферментативной активности.
- Генетический анализ. Учитывая разнообразие митохондриальных миопатий, для подтверждения диагноза обязательно выполняется секвенирование митохондриальной ДНК. При исследовании проверяется наличие мутаций, которые чаще всего провоцируют заболевание у детей.
- Биохимические исследования. Заподозрить мышечные поражения, связанные с нарушенным окислительным фосфорилированием, удается по увеличению показателей лактата и пирувата в крови, цереброспинальной жидкости.
Лечение митохондриальных миопатий
В клинической неврологии отсутствуют эффективные методы терапии патологии у детей. Суть медицинской помощи сводится к уменьшению моторного дефицита, своевременной коррекции осложнений, стимуляции обменных процессов в митохондриях. Наибольшую результативность демонстрируют следующие группы медикаментов:
- Аминокислоты. L-аргинин рекомендован неврологами в острой фазе для улучшения кровоснабжения мозга при осложнении миопатии инсультом, в резидуальном периоде болезни для предупреждения повторных приступов ишемии нервной ткани.
- Энерготропные препараты. Чтобы улучшить энергообеспечение тканей ребенка, эффективны препараты с левокарнитином, янтарной кислотой, коэнзимом Q10, широко используется комплекс витаминов группы В, аскорбиновая кислота, альфа-токоферол.
- Антиконвульсанты. При сочетании миопатии с эпилептическими пароксизмами показаны противосудорожные препараты группы сульфат-замещенных моносахаридов, бензодиазепинов, ограничено применяются барбитураты. Вальпроаты для лечения детей с митохондриальными нарушениями не назначаются.
Важным компонентом лечения у детей является нейродиетология, которая предполагает исключение веществ, оказывающих негативное влияние на обменные процессы (терапия «обхождения блока»). Рекомендована кетогенная диета, другие виды высокожировых диет. По показаниям проводится лечебное энтеральное или парентеральное питание, в тяжелых случаях устанавливается гастростома.
Для коррекции моторных нарушений применяется расширенный комплекс физиотерапии: ультразвуковая терапия, электромиостимуляция, электрофорез с ингибиторами ацетилхолинэстеразы. Хороший эффект демонстрирует лечебный массаж, индивидуально подобранный комплекс ЛФК. Чтобы избежать перегрузки ослабленных мышц, широко используются занятия в бассейне. Также требуется ортопедическая коррекция, подбор специальной обуви ребёнку .
Прогноз и профилактика
Поскольку митохондриальные миопатии пока являются неизлечимыми болезнями, прогноз неблагоприятный. Улучшить качество жизни пациентов удается с помощью комплексной реабилитации, однако при развернутой клинической картине смерть нередко наступает в детском или молодом возрасте. Для профилактики семейным парам с отягощенной наследственностью необходимо медико-генетическое консультирование при планировании беременности.
1. Митохондриальные болезни: миопатии, энцефаломиопатии и энцефаломиелополиневропатии/ В.М. Казаков, А.А. Скоромец, Д.И. Руденко, Т.Р. Стучевская// Неврологический журнал. — 2018. — №6.
3. Влияние дисфункции митохондрий на клинические проявления наследственных миопатий/ Д.А. Харламов, В.С. Сухоруков// Российский вестник перинатологии и педиатрии. — 2013. — №4.
4. Митохондриальные миопатии в сочетании с кардиомиопатией. Новые подходы к лечению/ О.С. Страхова, Ю.М. Белозеров, С.В. Перминов, В.В. Давыдкин// Альманах клинической медицины. — 2001. — №4.
Миотонический синдром у детей
Миотонический синдром у детей — это нервно-мышечная патология, для которой характерна затрудненная релаксация мускулатуры после ее напряжения. Встречается при наследственных генетических дефектах либо как проявление неврологических, эндокринных, метаболических, аутоиммунных заболеваний. Синдром характеризуется нарушениями двигательной активности, асимметрией мышц, задержкой психомоторного развития. Для диагностики необходим неврологический осмотр, инструментальные методы (ЭМГ, МРТ) и генетическое обследование. Лечение миотонии симптоматическое с применением физиотерапии, ЛФК и массажа, психолого-педагогической коррекции.
Частота встречаемости наследственных форм миотонического синдрома (МС) варьирует от 14 до 23 случаев на 100 тыс. детского населения. В отношении приобретенных вариантов статистических данных нет, поскольку в таких случаях патология не регистрируется как самостоятельная нозологическая единица и входит в структуру других неврологических болезней. Актуальность проблемы в педиатрической практике заключается в прогрессирующем течении заболевания, невозможности назначить этиотропное лечение и значительном ухудшении качества жизни детей при наследственных видах патологии.
В детском возрасте манифестируют врожденные формы, которые передаются преимущественно по аутосомно-доминантному, реже — аутосомно-рецессивному типу (синдром Шварца-Джампела, в части случаев — болезнь Томпсона). Самая частая форма наследственного МС — миотоническая дистрофия Россолимо-Штейнерта-Баттена-Куршмана, которая составляет до 97% всех генетических миотоний и связана с мутацией гена DMPK на 19-й хромосоме. Реже это заболевание обусловлено генетическими дефектами в 3-й (3q21) и 15-й (15q21-q24) хромосомах.
Вторичным миотоническим синдромом могут сопровождаться:
- родовые травмы с церебральными повреждениями;
- тяжелые формы перинатальной энцефалопатии;
- органические поражения ЦНС (опухоли, последствия перенесенных энцефалитов, ЧМТ);
- врожденные и воспалительные миопатии;
- миоплегии (болезнь Гамсторпа, периодический паралич);
- дефицитные состояния (рахит);
- эндокринные и метаболические расстройства (микседема, гипофункция паращитовидных желез, гиперкалиемия);
- аутоиммунные нарушения (приобретенная нейромиотония);
- длительное лечение клофибратом, калийсберегающими диуретиками, препаратами калия.
В механизме развития заболевания ведущим является нарушение процесса расслабления мышцы в результате метаболического или ионного дисбаланса. При этом изменяются свойства мембран миоцитов, происходит их усиленная деполяризация и повышение возбудимости. При совершении определенного действия с участием мускулатуры миофибриллы долго не могут расслабиться и вернуться в исходное положение, вследствие чего и возникает феномен миотонической задержки.
Лучше всего изучен патогенез дистрофической миотонии 1 типа, связанной с нарушениями на уровне экспрессии нуклеотидного повтора CTG. У детей изменяется концентрация миотонин-протеинкиназы DMPK, которая присутствует в скелетной мускулатуре, миокарде и ЦНС. В редких случаях клинические проявления обусловлены дисфункцией ионных каналов клеточных мембран, которая отмечается при аутоиммунных нарушениях.
Симптомы
Главный признак миотонического синдрома — скованность мышечных групп в конце движения. Это проверяется с помощью симптома «кулака»: после сжатия пальцев ребенок несколько секунд не может разжать кулак, и для этого ему нужно прилагать много усилий. Синдром характеризуется затруднениями жевания, неспособностью быстро открыть глаза после зажмуривания. Для наследственных форм МС у детей типично усиление симптоматики при целенаправленных движениях, в холодное время года.
При врожденных формах миотонического синдрома возможно асимметричное развитие мускулатуры. Если патология проявляется с первых месяцев жизни, родители замечают, что ребенок не способен удерживать голову, слабо сосет грудь, у него хуже сформированы рефлексы. Для грудничков с миотоническим синдромом характерно запоздалое становление моторики. В раннем детстве наблюдается повышенная утомляемость малыша, отсутствие интереса к активным играм, отставание в физическом развитии.
На поздних стадиях заболевания у детей происходит атрофия мышц, поэтому типичные признаки миотонического синдрома ослабевают, а потом исчезают. В клинической картине преобладает выраженная мышечная слабость, особенно в дистальных мышцах конечностей. Это обуславливает изменение походки, приводит к появлению вялых парезов и параличей. Поражение лицевых мышц проявляется постоянно «печальным» выражением лица и бедностью мимики.
Врожденные миотонические синдромы относят к мультисистемным поражениям, поэтому у таких детей зачастую диагностируют сопутствующие сердечно-сосудистые, неврологические и эндокринные нарушения. Особую опасность представляет нарушение иннервации дыхательной мускулатуры, из-за чего у пациентов нарастать дыхательная недостаточность вплоть до асфиксии. Церебральные симптомы, как правило, сочетаются с когнитивными нарушениями.
Лечение наследственных разновидностей миотонического синдрома затруднено, поэтому нередки летальные исходы в молодом возрасте. До 80% смертей вызваны вторичной пневмонией на фоне аспирации пищи и нарушений глотания, жизнеугрожающими аритмиями, которые развиваются вследствие дегенерации проводящей системы сердца. У детей может возникать глубокая умственная отсталость, приводящая к инвалидности.
При физикальном осмотре детский невролог проверяет симптом «кулака», оценивает сохранность рефлексов, мышечную силу. На основании полного физикального осмотра специалисту удается поставить предварительный диагноз. Чтобы подтвердить наличие и выяснить причины миотонического синдрома, назначаются следующие диагностические исследования:
- Электромиография. Патогномоничный признак миотонии на ЭМГ — появление высокоамплитудных разрядов со звуковым феноменом при использовании игольчатых электродов. При прогрессировании болезни присутствуют симптомы миопатии: снижение амплитуды потенциалов, полифазные двигательные единицы.
- Биопсия мышц. На ранних стадиях в биоптате определяются неспецифические миопатические изменения: центрально расположенные ядра, уменьшение длины волокон, признаки денервации — наличие пикнотических ядерных глыбок и маленьких угловых мускульных волокон.
- Магнитно-резонансная томография. По результатам МРТ конечностей врач оценивает степень развития мускулатуры и замещения ее жировой тканью. Учитывая сопутствующее поражение ЦНС, рекомендована МРТ головного мозга для обнаружения структурных изменений белого вещества и желудочков.
- Генетическая диагностика. Исследование генома для выявления типичной мутации — «золотой стандарт» для постановки диагноза врожденных вариантов миотонического синдрома у детей. По показаниям можно проводить тестирование в антенатальном периоде, если в семьях родителей были случаи миотонии.
Лечение миотонического синдрома у детей
Консервативная терапия
Этиопатогенетическое лечение миотонического синдрома пока не разработано, поэтому в детской неврологии ограничиваются симптоматическими средствами. Медикаменты подбираются с учетом конкретной ситуации и выраженности патологических изменений. Для замедления прогрессирования проводится лечение противосудорожными средствами и миорелаксантами, для подавления аутоиммунных реакций назначают глюкокортикоиды.
Решающее значение в терапевтической схеме отводится упражнениям ЛФК, которые укрепляют мышечный корсет, помогают сбалансировать развитие разных групп мышц, что улучшает качество жизни больных детей. Показано физиотерапевтическое лечение в виде электрофореза с кальцием, электростимуляции мышц, бальнеотерапии. Для уменьшения миотонического синдрома полезен массаж.
Реабилитация
Учитывая серьезные двигательные нарушения, лечение дополняется регулярными занятиями в реабилитационных центрах с использованием тренажеров и индивидуальных методик. Нарушения артикуляции у детей корректируются занятиями с логопедом. При задержке умственного развития ребенку может потребоваться помощь олигофренопедагога либо обучение в специализированной школе.
Если вовремя начать лечение, при приобретенных формах удается восстановить мышечную активность. Наследственный миотонический синдром отличаются сомнительным прогнозом. Средняя продолжительность жизни пациентов составляет 35 лет при раннем дебюте болезни. Специфические меры профилактики миотонического синдрома не разработаны. Семьям с отягощенным анамнезом требуется медико-генетическое консультирование.
1. Клинический случай дистрофической миотонии 1-го типа/ Н.В. Ноздрюхина, А.А. Струценко, Е.Н. Кабаева// Трудный пациент. — 2019.
2. Миотоническая дистрофия. Современное представление и собственное наблюдение/ Т.И. Стеценко// Современная педиатрия. — 2014.
Миопатии
Миопатии — группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Причины миопатий
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр.), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и др.), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и др. формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные - возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Симптомы миопатий
Большинство миопатий имеют постепенное начало с появления небольшой мышечной слабости в конечностях, более быстро возникающей усталости от ходьбы и другой физической нагрузки. В течение нескольких лет происходит нарастание слабости, появляются и прогрессируют мышечные атрофии, возникают деформации конечностей. Из-за значительной мышечной слабости пациенты с трудом поднимаются с пола и ходят по лестнице, не могут прыгать и бегать. Для того, чтобы встать со стула, им приходится использовать специальные приемы. Характерен вид больного: крыловидно отстоящие лопатки, опущенные плечи, выпяченный вперед живот и усиленный поясничный лордоз. Наблюдается «утиная походка» — пациент передвигается, раскачиваясь в стороны.
Патологические изменения при миопатиях происходят симметрично в мышцах конечностей и туловища. Как правило, мышечные атрофии наблюдаются в проксимальных отделах рук и ног. В связи с этим мышцы дистальных отделов конечностей могут выглядеть гипертрофированными. Такая миопатическая псевдогипертрофия наиболее заметна в мышцах голеней. Наряду с нарастанием мышечной слабости наблюдается постепенное угасание сухожильных рефлексов и прогрессирующее снижение мышечного тонуса, т. е. развивается и усугубляется периферический вялый паралич. Со временем результатом резкого ограничения активных движений становятся контрактуры суставов.
Миопатии могут сопровождаться поражением мимических мышц, что проявляется невозможностью вытянуть губы трубочкой, свистеть, нахмурить лоб или улыбнуться. Поражение круговой мышцы рта приводит к появлению дизартрии, связанной с затруднением произношения гласных звуков.
Клиника некоторых миопатий включает поражение дыхательной мускулатуры, приводящее к возникновению застойной пневмонии и развитию дыхательной недостаточности. Возможны патологические изменения сердечной мышцы с возникновением кардиомиопатии и сердечной недостаточности, мышц глотки и гортани с развитием дисфагии и миопатического пареза гортани.
Особенности отдельных форм миопатии
Ювенильная миопатия Эрба наследуется аутосомно-рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече-лопаточно-лицевая миопатия Ландузи - Дежерина имеет аутосомно-доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия — аутосомно-доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно-доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов - уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и др. ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение миопатий
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии и т. д.
Наиболее неблагоприятны в прогностическом плане наследственные миопатии, проявляющиеся в раннем детском возрасте. В остальном прогноз зависит от формы миопатии, вовлеченности в процесс сердечной и дыхательных мышц. Прогноз вторичных миопатий более благоприятный при условии успешного лечения основного заболевания.
Профилактикой первичных миопатий служит тщательный сбор семейного анамнеза и обязательное консультирование у генетика пар, планирующих беременность. Профилактикой вторичных миопатий является исключение токсических воздействий на организм, своевременное лечение инфекционных и эндокринных заболеваний, коррекция метаболических нарушений.
Идиопатические воспалительные миопатии
Дерматополимиозит - гетерогенная группа хронических воспалительных заболеваний с преимущественным поражением поперечно - полосатой и гладкой мускулатуры с нарушением двигательной функции, кожи в виде эритемы и отека, с частым поражением внутренних органов, относящаяся к диффузным болезням соединительной ткани.
Код протокола:
- Поражение скелетных мышц (прогрессирующая симметричная слабость проксимальной группы мышц верхних и нижних конечностей, сгибательных мышц шеи, отек мышц, поражение мышц гортани, глотки, верхней трети пищевода)
- Поражение кожи (параорбитальная «гелиотропная» сыпь, эритема Готтрона, фотодерматит, кожный зуд, панникулит, «рука механика»)
Факторы риска: Этиология полимиозита/дерматомиозита не известна. Генетическая предрасположенность у монозиготных близнецов и кровных родственников больных, косвенно- инфекционные факторы. Носительство HLA - B8/DR3, HLA- B14, HLA - B40 антигенов гистосовместимости.
• Кожный синдром: эритема, имеющая вид солнечного ожога или пурпурно-лиловая на открытых частях тела, над суставами, параорбитальный отек, эритема верхнего века с лиловым оттенком - «дерматомиозитовые очки», капилляриты ладоней, пальцевых подушечек; плотный или тестоватый отек лица, кистей, реже стоп, голеней, туловища (в сочетании с эритемой).
• Скелетно-мышечный синдром: генерализованное поражение поперечно-полосатых мышц, на ранних этапах - нарастающая слабость мышц плечевого пояса и проксимальных отделов нижних конечностей, миалгии, отеки мышц; позже миосклероз, контрактуры, атрофии проксимальных отделов конечностей.
• Висцерально-мышечный синдром: поражение дыхательных мышц, включая диафрагму (одышка, высокое стояние и вялость дыхательных экскурсий диафрагмы, снижение жизненной емкости легких и резерва дыхания), глотки,пищевода, гортани (дисфагия, поперхивание, дисфония), миокарда (миокардит, дистрофия, интерстициальный отек).
• Лабораторные данные:креатинурия, повышение содержания в крови трансаминаз, миоглобина, альдолазы, креатининфосфокиназы, лактатдегидрогеназы.
• Морфологическая картина: воспалительно-дистрофические изменения, которые заканчиваются развитием склероза, атрофией мышечных волокон, кальцинозом; дистрофия мышечных волокон характеризуется разволокнением, потерей поперечнополосатой исчерченности, истончением волокна, глыбчатым распадом, гомогенизацией и постепенным их исчезновением, фрагментацией мышечных волокон вплоть до некроза; воспалительная реакция проявляется периваскулярным отеком, очагами круглоклеточной инфильтрации лимфоцитов и плазматических клеток; воспалительная инфильтрация располагается периваскулярно между мышечными волокнами и межмышечной соединительной тканью; мышечные волокна отечны, разъединены.
Диагноз «дерматомиозит» достоверен при наличии двух-трех признаков, причем обязательны кожный и мышечный синдромы, которые наиболее типичныи в 100% случаев являются первыми признаками болезни. С висцеральной патологии дерматомиозит практически не начинается.
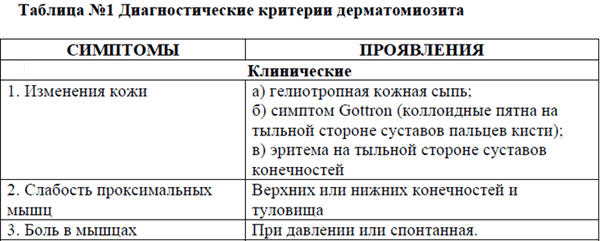
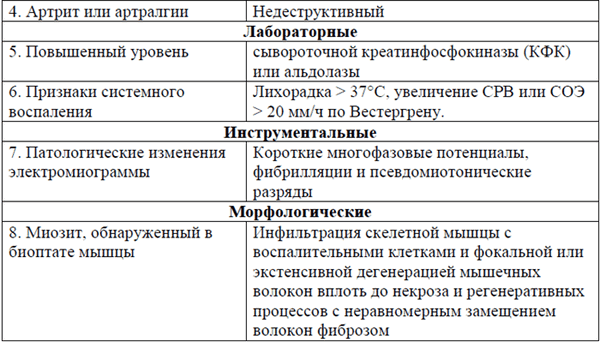
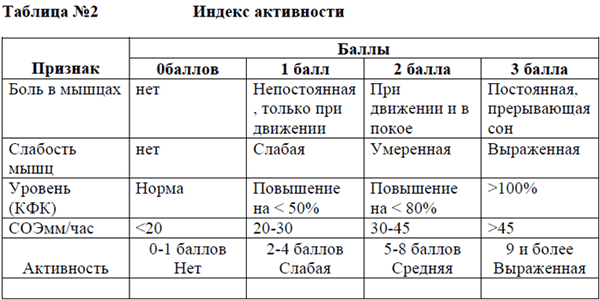
Наличие по крайней мере одного типа поражение кожи и по крайней мере 4 признаков (из пунктов 2-8) соответствуют диагнозу дерматомиозит. Наличие по крайней мере 4 признаков (из пунктов 2-8) соответствуют диагнозу полимиозит.
Дерматомиозит и полимиозит с миозитспецифическими антисинтетазными Jo-1-антителами имеют следующие особенности:
Дерматомиозит (полимиозит) с миозитспецифическими антителами неантисинтетазными цитоплазматическими анти-SRP имеет следующие особенности:
Дерматомиозит (полимиозит) с миозитспецифическими антиядерными антителами - анти-РМ наиболее характерен для перекрестного синдрома - сочетания полимиозита и системной склеродермии. Иногда эти антитела выявляются при ювенильном дерматомиозите.
• редкая ассоциация с диффузными болезнями соединительной ткани и злокачественными новообразованиями;
• в биоптатах мышц выявляются «очерченные вакуоли», крупные внутриядерные и внутриплазматические включения (при световой микроскопии) и микротубулярные элементы (при электронной микроскопии).
Изменения кожи, лихорадка, слабость проксимальных мышц, боль в мышцах, боли в суставах,в спине, приступы кашля, затрудненное глотание. Основные принципы сбора анамнеза у больного с подозрением на полимиозит/дерматомиозит включают выяснение обстоятельств заболевания.
• Общий анализ крови: У части больных признаки умеренной анемии, лейкоцитоз с нейтрофильным сдвигом влево, реже - лейкопения, эозинофилия, СОЭ увеличивается соответственно активности патологического процесса.
• Биохимический анализ крови: повышение содержания альфа2 и у-глобулинов, серомукоида, фибрина, фибриногена, сиаловых кислот, миоглобина, гаптоглобина, креатина, активности креатинфосфокиназы (нормальный уровень КФК при тяжелой мышечной атрофии и при наличии в крови ингибитора КФК), трансаминаз, особенно АсАТ, ЛДГ и альдолазы, что отражает остроту и распространенность поражения мышц.
• Иммунологические исследования крови: снижение титра комплемента, в небольшом титре РФ, в небольшом количестве и незакономерно - LE-клетки, антитела к ДНК, снижение количества Т-лимфоцитов и Т-супрессорной функции, повышение содержания IgM и IgG и снижение - IgA; HLA B8, DR3, DR5, DRW52, высокие титры миозитспецифических антител.
• Исследование биоптатов кожно-мышечного лоскута: тяжелый миозит, потеря поперечнойисчерченности, фрагментация и вакуолизация мышц, круглоклеточная инфильтрация, атрофия и фиброз их. В коже - атрофия сосочков, дистрофия волосяных фолликулов и сальных желез, изменения коллагеновых волокон, периваскулярная инфильтрация.
• Электромиограмма: тяжелые мышечные изменения - короткие волны с полифазовыми изменениями, фибриллярные осцилляции в состоянии покоя.
• Рентгенологическое исследование способствует уточнению степени поражения мягких тканей и внутренних органов. Рентгенограммы следует проводить с помощью мягкого излучения, чтобы получить структуру мягких тканей. В острой стадии дерматомиозита мышцы выглядят более прозрачными, отмечаются просветления. При хроническом дерматомиозите появляются кальцификаты в мягких тканях. В легких определяется интерстициальный фиброз, преимущественно базальных отделов, кальцификаты плевры. Сердце увеличено в размерах. В костях может быть умеренный остеопороз.
2.Биохимические исследования:общий белок, СРБ, РФ,креатинин, липидный спектр,глюкоза, КФК, альдолаза, лактатдегидрогеназа, АЛТ, АСТ, билирубин, тимоловая проба
3.Иммунологические исследования:АНФ, антитела к нативной и денатурированнойДНК, миозитспецифические антисинтетазные антитела (к гистидин-синтетазе (анти-Jo-1).
5.Определение СА-15,3, СА-125, PSA (исключение рака яичников, молочной железы, предстательной железы)
Однократно: Рентгенография органов грудной клетки, кистей, УЗИ почек, печени, селезенки, ЭКГ, ЭХОКГ, электромиография, фиброгастроскопия. Биопсия проксимальной мышцы (дельтовидной, ягодичной).
Дополнительные инструментальные и лабораторные исследования проводятся в зависимости от наличия сопутствующей патологии, наличия висцеропатий и осложнений медикаментозной терапии.
Дифференциальную-диагностику ИВМ проводят с широким кругом заболеваний, сопровождающихся проксимальной мышечной слабостью.
- неврогенные миопатии (амиотрофический склероз, полиневропатия, спинальная амиотрофия Кугельберга-Веландера, синдром Кеннеди (спинобульбарная мышечная атрофия передних рогов спинного мозга), демиелинизирующие полинейропатии (острая, хроническая), невральная перонеальная амиотрофия Шарко-Мари-Тута).
- Первично-мышечные заболевания: инфекционные миозиты (бактериальные и вирусные миозиты), поражение мышц при токсоплазмозе, трихинеллезе, цистицеркозе, эхинококкозе
- лекарственные миопатии могут возникать при использовании ГК, пеницилламина, хлорохина (например, делагил), гидроксихлорохина (например, плаквенил), колхицина, статинов, гемфиброзила, эритромицина, эметина, зидовудина, а также при алкогольной и наркотической (кокаин) интоксикации, длительном приѐме гормонов щитовидной железы в высоких дозах. Для стероидной миопатии характерны нормальный уровень КФК, увеличение мышечной силы на фоне снижения дозы ГК
- метаболические миопатии (нарушение метаболизма гликогена, липидов, пуринов). Характерный признак-снижение толерантности к физической нагрузке и восстановление мышечной силы на фоне отдыха.
• Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с благоприятным прогнозом.
• Адекватная инициальная доза: в зависимости от тяжести заболевания начальная доза колеблется от 1 до 2 мг/кг/сут. • Ежедневный прием ГК.
Суточную дозу ГК в начале лечения следует делить на 3 приема (оценивая ее переносимость), однако, в течение первой половины дня; затем перевести пациента на прием полной дозы ГК в утренние часы. Оценка эффективности терапии проводиться через 2-4 недели от начала терапии ГК. Положительный эффект терапии расценивается при начавшемся снижении уровня КФК, АСТ, АЛТ, уменьшении интенсивности кожных проявлений, нарастании мышечной силы. В случае отсутствия положительной динамики - увеличить дозу ГК до 1,5 мг/кг/сут. Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
Снижение дозы ГК начинается при нормализации уровня КФК в сыворотке крови, исчезновении спонтанной активности при и-ЭМГ, нарастании мышечной силы, объема
движений и проводиться под строгим клинико-лабораторным контролем. Доза ГК постепенно снижается по ¼ дозы от исходной в месяц, в среднем, по ½ - ¼ таблетки в 5-7-10 дней до достижения поддерживающего уровня. Темп снижения зависит от исходной дозы ГК и степени активности болезни. Чем ниже доза ГК, тем медленнее ее снижение.
Поддерживающая доза ГК индивидуальна: 5-10, реже 15 мг/сутки и зависит от клинико-иммунологического подтипа болезни, возраста больного. При ЮДМ известны случаи клинико-лабораторной ремиссии на фоне длительной отмены терапии. Полная отмена ГК у взрослых пациентов, как правило, ведет к обострению болезни, даже если они несколько лет находились в состоянии полного клинического ответа.
Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и не служит поводом для применения меньших (не адекватных) доз ГК назначаемых внутрь, как в острый период болезни, так и при ее обострении.
стью которых является заведомо «плохой ответ» на терапию ГК: АСС c фиброзирующим альвеолитом, у пациентов антител к SRP
⎯ Тяжелые побочные эффекты ГК, лимитирующие назначение адекватной дозы ГК (неконтролируемые сахарный диабет или артериальная гипертензия, острая язва желудка, множественные остеопоретические переломы).
1. Наиболее тяжелым и недостаточно контролируемым монотерапией ГК при ПМ/ДМ синдромом является АСС, маркеруемый выявлением миозит-специфических антисинтетазных антител (анти Jo-1, анти PL-7, анти PL-12 и др.) в сыворотке крови. Плохой прогноз определяется вовлечением в патологический процесс легочной ткани - ИПЛ с развитием фиброзирующего альвеолита.
2. Объем терапии и выбор препарата (в сочетании с ГК) определяется тяжестью ИПЛ (по данным КТ и функциональных легочных тестов - форсированной жизненной емкости легких (ЖЕЛ), диффузионной способности легких (DLCO) и с учетом анамнеза (ранее применяемые иммуносупрессивные препараты).
3. Основное место в лечении ИПЛ занимает циклофосфамид (ЦФ), назначаемый внутривенно в дозе 500 мг/м2 -750 мг/м2 мг в месяц в сочетании с ГК.
5. Контроль эффективности ЦФ осуществляется по динамической оценке (1 раз в 6 месяцев) форсированной ЖЕЛ, показателей DLCO, а также данных КТ легких.
6. При агрессивном течении СФА при выраженном снижении ЖЕЛ и DLCO, а также, в случае неэффективности ранее применяемой терапии ЦФ, целесообразно применение ритуксимаба.
1. Нарушение глотания (дисфагия) является фактором риска аспирационной пневмонии, течение и терапия которой осложняется иммуноскомпроментированностью пациентов, связанной с терапией высокими дозами ГК и цитостатиков.
2. Рекомендовано проведение пульс-терапии ГК (метипред 1000мг) N 3 в сочетании с пероральным приемом ГК в адекватной дозе. 3. Тяжелая дисфагия
Наличие язвенно-некротического васкулита является показанием для проведения пульс-терапии циклофосфамидом в дозе 600-800-1000 мг в месяц в сочетании метилпреднизолоном 500-1000мг.
Кожный синдром при ДМ в сочетании с проксимальной мышечной слабостью отражает активность болезни и, как правило, контролируется ГК в адекватных дозах в острый период болезни. При резистентном кожном синдроме, сохраняющемся на фоне восстановления мышечной силы, рекомендуется применение антималярийных препаратов (гидроксихлорохин по 200-400 мг/сут), топических стероидов.
1. Наличие артрита при ПМ/ДМ может присутствовать в начале болезни. Артриты входят в состав симптомокомплексаАСС, хорошо контролируются ГК и не требуют дополнительного лечения. 2. Сгибательные контрактуры, как правило, локтевых, реже коленных суставов, развиваются в острый период ПМ/ДМ и обусловлены воспалительным поражением мышечной ткани, а не непосредственным поражением суставов. Дополнительного медикаментозного лечения не требуется.
1. Метотрексат по 7,5-25 мг/нед внутрь или внутривенно (при недостаточной эффективности или плохой переносимости перорального приема препарата, особенно в высоких дозах).
2.Азатиоприн по 2-3 мг/кг/сут (100-200 мг/сут) 3.Циклоспорин А (Сандиммун) по 2,5-5.,0 мг/кг/cутки назначают пациентам с резистентными к ГК формами заболевания, в т.ч. при хроническом течении болезни, связанной с неадекватно малой инициальной дозой ГК.
4. Микофенолатамофетил (ММФ). Прием начинают с дозы 1000 мг/сут (в 2 приема), постепенно титруя дозу до 2000 мг/сут под контролем показателей общего и биохимического анализов крови.
Применение внутривенного иммуноглобулина (ВИГ) 2 г/кг 1 раз в месяц в течение 3 месяцев является эффективным методом лечения ПМ/ДМ (особенно ЮДМ), резистентного к стандартной терапии. Потенциальным показанием для ВИГ является тяжелая дисфагия.
Плазмаферез следует использовать главным образом у больных с тяжѐлым, резистентным к другим метода лечения ПМ/ДМ в сочетании с ГК и цитотоксическими препаратами.
1. Глюкокортикостероидные препараты (обязательно в сочетании с препаратами кальция и витамином D): Преднизолон 20 - 30 мг/сутки в 3 приема 1 неделя,
затем в 1 прием не менее 2 мес. после достижения ремиссии, с последующим снижением по 1,25 мг (1/4 т) в 2 недели до минимальной поддерживающей дозы 10-15 мг/сутки на срок, не менее двух лет.
3. Симптоматическая терапия в зависимости от сопутствующей патологии и осложнений медикаментозной терапии.
При средней степени активности
1. Глюкокортикостероидные препараты (обязательно в сочетании с препаратами кальция и витамином D): Преднизолон 1 мг/кг/сутки в 3 приема 1 неделя, затем в 1 прием не менее 2 - 3 мес. после достижения ремиссии, с последующим снижением по 1,25 мг (1/4 т) в неделю до минимальной поддерживающей дозы 15-20 мг/сутки на срок, не менее двух лет.
3. Симптоматическая терапия в зависимости от сопутствующей патологии и осложнений медикаментозной терапии.
При высокой степени активности
1.Пульс терапия: 1г Метилпреднизолон + физиологический раствор 100-200 мл + Гепарин 10000ЕД в/в капельно3дня подряд.При ювенильном дерматомиозите,при рефрактерных формах дерматомиозита в сочетании с плазмаферезомс последующим переходом на пероральный прием кортикостероидов.
2.Глюкокортикостероидные препараты (обязательно в сочетании с кальция и витамином D): Преднизолон 1 - 2 мг/кг/сутки в 1 - 2 приема не менее 1 мес. после достижения ремиссии, с последующим снижением по 1,25 мг (1/4 т) в неделю до минимальной поддерживающей дозы 25-35 мг/сутки на срок, не менее двух лет.
7.Генно-инженерно-биологическая терапия - инфликсимаб 3мг/кгпо схеме при тяжелом, резистентном к стандартной терапии течении заболевания, ритуксимаб - 5мг/кг по схеме.
8. Симптоматическая терапия в зависимости от сопутствующей патологии и осложнений медикаментозной терапии.
- Метилпреднизолон(Солу-Медрол ) 250 мг, 500мг, 1000мг, порошок для приготовления инъекционного раствора
Читайте также:
- Техника ламинэктомии при опухоли грудного позвонка
- Диагностика эпидемического паротита. Лечение эпидемического паротита. Профилактика эпидемического паротита.
- Влияние скорости на дорожный травматизм. Превышение скорости как причина травматизма
- Гормональные рилизинг-системы: кольцо, пластырь, имплант
- Искусственные заболевания. Особенности искусственных заболеваний