Синдром Нишамена (Nichamin) - синонимы, авторы, клиника
Добавил пользователь Skiper Обновлено: 28.01.2026
Среди заболеваний печени, протекающих в хронической форме, особое место занимает синдром Жильбера или синдром Печорина. Патология, вызываемая на генетическом уровне, не провоцирует цирроз либо рак. Она появляется из-за большой доли в кровяной сыворотке непрямого билирубина. При ней растет риск возникновения не только желчнокаменной болезни, но и холестаза. Также обострения заметно снижают качество жизнедеятельности больного. С наличием такого заболевания нельзя брать в армию.
Для результативного лечения, профилактики рассматриваемого синдрома важно своевременно обращаться к гастроэнтерологам клиник МЕДСИ. Врачи оперативно выявят признаки заболевания, назначат диагностические мероприятия для подтверждения диагноза, подготовят максимально эффективный лечебный курс.
Определение синдрома Жильбера
Болезнь Жильбера относится к наследственным патологиям, поэтому называется семейной доброкачественной гипербилирубинемией, сопровождает человека всю жизнь. Хронический характер протекания приводит к чередованию ремиссий с эпизодами желтухи, вызванной неблагоприятными факторами.
Непрямая гипербилирубинемия проявляется в подростковом возрасте при половом созревании. У представительниц женского пола она диагностируется в 3. 4 раза реже, нежели у мужчин.
Причины возникновения синдрома Жильбера
Обмен билирубина нарушается из-за стойкого видоизменения гена UGT1A1, кодирующего фермент уридиндифосфат-глюкуронилтрансферазу, который необходим гепатоцитам для расщепления билирубина.
Желчный пигмент, именуемый билирубином, подразделяется на непрямой, прямой тип. Первая разновидность в значительных дозах токсична, накапливается в кожном покрове, окрашивая кожу в характерный желтый цвет.
Непрямой билирубин в крови появляется при разрушении эритроцитов. У взрослых красные кровяные тельца живут 120 суток. Затем они заменяются новыми клетками крови. При разрушении старого эритроцита происходит высвобождение непрямого билирубина, который связывается с альбумином, доставляется в печень.
В органе, избавляющем кровь от токсинов, желчный пигмент в гепатоците трансформируется в прямой билирубин посредством уридиндифосфат-глюкуронилтрансферазы, направляется в просвет кишечника вместе с желчью. В результате осуществляется обезвреживание и выведение вредоносного билирубина.
Классификация
Подвидов у синдрома Жильбера нет. Лечащему медперсоналу важно исключить иные разновидности желтухи, относящиеся к симптоматике более опасных заболеваний, чем семейная доброкачественная гипербилирубинемия.
Желтушный криз классифицируется по причине на следующие виды:
- Печеночный
- Механический
- Гемолитический
Болезнь причисляется к печеночному типу, потому что обусловлена отклонениями в функционировании печени при удалении билирубина.
Симптомы синдрома Жильбера
Основными признаками считаются пожелтения слизистых оболочек, кожного покрова, глазных склер. Желтизна бывает с различной насыщенностью, проявляется волнообразно.
Эпизодические обострения провоцируются разнообразными факторами - как внутренними, так и внешними. Больной может чувствовать в правом подреберье тяжесть, боль. Малоприятные ощущения связаны с неблагоприятным изменением состава желчи, который и приводит к нарушению ее оттока. В итоге фиксируются отклонения в пищеварении: меняется характер стула, усиливается газообразование, появляется отрыжка. Иногда регистрируется головокружение, ухудшение сна, подавленность настроения, быстрое наступление утомления.
К каким врачам обращаться?
Изначально посещается педиатр либо терапевт. Медработник всесторонне обследует, исключит иные разновидности болезней. Благодаря полученным результатам исследований, эксперт направит к гастроэнтерологу, генетику, а также гепатологу, который диагностирует и лечит патологии печени, желчевыводящих путей.
Диагностика синдрома Жильбера
В первую очередь кровь анализируется на общий билирубин, доля которого в одном литре варьируется в пределах 21. 51 микромоля при синдроме Жильбера. Физическое напряжение либо заболевание периодически повышает его количество до уровня, равного 85. 140 мкмоль/л.
Подозревая наличие болезни Жильбера, назначается одна из трех проб (анализов):
- С голоданием. За счет полноценного голодания или строгого соблюдения низкокалорийного рациона в течение пары суток, предполагающего каждодневное употребление еды, содержащей не более 400 килокалорий, доля билирубина в кровяной сыворотке увеличивается на 50. 100%. Первый раз концентрация компонента желчи уточняется утром натощак в день старта пробы. Второе определение количества выясняется по прошествии двух суток
- С фенобарбиталом. Лекарственный препарат активизирует фермент глюкуронилтрансферазы, понижает концентрацию билирубина
- Генетическое исследование. Отличается максимальной точностью, поскольку позволяет выявить мутацию соответствующего гена
Методы лечения
Отсутствует не только этиотропная, но и патогенетическая лечебная терапия. Современная медицина способна лишь облегчить либо снять симптоматику при помощи следующих методик:
- Избавление от таких факторов влияния как инфицирование, физические или психические перегрузки. Стоит исключить употребление спиртосодержащих жидкостей, лекарственных средств, токсичных для печени
- Прекращение приема лекарств, способствующих глюкуронированию, отделению альбумина от билирубина. К ним причисляются салицилаты с сульфаниламидами, гепатрин и пероральные контрацептивы
- Отведение прямого билирубина посредством обильности питья для наращивания выработки мочи, активированного угля, адсорбирующего в кишечном тракте билирубин
- Связывание альбумином уже имеющегося билирубина. Альбумин дозируется из расчета грамм на килограмм веса тела в течение одного часа. Особенно важна процедура перед обменным переливанием крови
- Расщепление фототерапией билирубина, находящегося в тканях. Применяются лампы синего свечения, имеющие длину волны на уровне 450 нм. Источники света должны размещаться от кожного покрова на дистанции в пределах 40. 45 см
- Исключение попадания большого объема лучей солнца
- Витаминотерапия. Стремятся насытить витаминами группы B
- Строгое соблюдение диеты, лимитирующей потребление продовольствия, содержащего консерванты с насыщенными жирными кислотами. Контроль питания
- Употребление желчегонных веществ
- Терапия заболеваний желчевыводящих путей хронического характера и таких же инфекций
- Переливание крови обменного типа. Используется в критических ситуациях
- Прием на основе назначенного курса таких гепатопротекторов, как экстракты артишока полевого, плодов расторопши, а также комбинированных протекторов растительного происхождения
Если фенобарбитал провоцирует вялость с сонливостью, нарушение координации телодвижений, то выбирается 0,05 грамма, принимаемые перед отходом ко сну. Это нужно для увеличения периода его употребления.
Когда самочувствие плохое при концентрации билирубина в крови на уровне 50 мкмоль/л, тогда назначается короткий лечебный курс с применением фенобарбитала. Ежесуточно принимается 0,03. 0,2 грамма в течение от пары недель до месяца. Допускается пользоваться валокордином, корвалолом, а также барбовалом, поскольку в них содержится фенобарбитал. Троекратно в сутки употребляется по 20. 25 капель. Следует отметить, что не каждому данный вариант поможет. Разрешается два-три раза в сутки использовать и кордиамин на протяжении 7 дней, отмеряя не более 30. 40 капель.
Терапию зиксорином организм переносит хорошо. Побочных явлений не наблюдается. В России с 1998 года запрещено смывать данное средство, поэтому флумецинол не изготавливается.
Страдающим синдромом Жильбера приходится сталкиваться и с желчнокаменной болезнью, а также холециститом. Исключается подобное развитие событий приемом отваров из желчегонных трав. Эксперты рекомендуют на регулярной основе очищать печень тюбажами с сорбитом либо ксилитом, пользоваться солью «Барбара» и карловарской.
Осложнения
Чаще всего образуются желчные камни, потому что в желчи оказывается мало соответствующих пигментов. Камни способны формировать пролежни в желчных протоках, которые могут разрываться. Появляются и свищи из-за них, поэтому показано консультирование у хирурга.
Профилактика
При синдроме Жильбера у взрослого человека или ребенка целью профилактических процедур является предупреждение развития желчнокаменной болезни, желтухи. Нельзя физически перегружаться. Запрещается кушать консервированное продовольствие, жареные блюда, жирную пищу, много пряностей.
Желтуха провоцируется и некоторыми медикаментами. Лечащий врач совместно с больным уточняет необходимость употребления подобных лекарств. Нередко польза от них перевешивает вероятность появления желтушного криза.
Инфекции, протекающие с лихорадкой, также способны привести к желтухе. Рекомендуется вовремя вакцинироваться, закаливаться, блюсти личную гигиену, выполнять общеукрепляющие мероприятия.
Зачастую синдром Жильбера протекает в тяжелой форме, если уже имеются заболевания печени. Благодаря своевременным методикам диагностирования вирусных гепатитов, алкогольной болезни печени, их лечения реально минимизировать риски, снизить тяжесть последствий.
В качестве профилактической меры требуется периодически проводить скрининговые обследования вместе с выполнением биохимического анализа крови. Исследования позволяют на ранней стадии выявлять желчнокаменную болезнь, считающуюся основным осложнением. При возникновении необходимости осуществляется ультразвуковое обследование органов, расположенных в брюшной полости. Благодаря раннему старту консервативной лечебной терапии, удается предупредить формирование желчных камней, избежать хирургического вмешательства.
Преимущества обращения в МЕДСИ
Основные достоинства, которые заставляют многих людей с синдромом Жильбера обращаться за услугами именно нашей клиники:
- Высокий уровень медицинского обслуживания. Используется только современное оборудование, которое зарекомендовало себя только с положительной стороны во всем мире
- Ведущие специалисты. Врачи имеют большой опыт работы в данном направлении. Постоянно повышают уровень квалификации
- Комфортабельный стационар. В центре созданы все условия для нахождения в приятной и удобной обстановке. Вы будете чувствовать себя как дома
- Возможность плановой госпитализации по ОМС. Это весьма выгодно для пациента, работаем в различных профильных направлениях
- Допускается экстренная госпитализация. При наличии явных симптомов возможно быстрое оформление в стационар
- Уникальные программы подготовки перед госпитализацией. Вы можете быть уверены, что лечение в клинических условиях окажется эффективным.
Вопрос/ответ
Если возникла желтизна на кожном покрове и в районе глаз, это в любом случае плохо?
Нет. Желтуха, возникающая ввиду наличия данного синдрома, не всегда указывает на гибель клеток печени или воспалительный процесс. Исключена вероятность образования рака и цирроза.
В ходе развития синдрома Жильбера на генетическом уровне снижается активность фермента, участвующего при обезвреживании билирубина (примерно на 35%). Без его наличия билирубин не будет выводиться. Он как бы ждет своей очереди, оставаясь в составе крови.
Речь идет о наследственности, но ребенок первоначально был абсолютно здоров. Почему появилась желтизна, диагностировали синдром Жильбера?
Подростковый возраст сопровождается повышением концентрации половых гормонов. Такая особенность способна повлиять на обмен билирубина. Именно во время полового созревания его объем увеличивается.
В ходе генетического анализа не выявлено мутаций, характерных синдрому Жильбера. Гастроэнтеролог при этом не исключает его наличие. Почему?
Чаще всего у европеоидного человека, у которого диагностировали синдром Жильбера, присутствует гомозиготный полиморфизм на двух основаниях с последовательностью TATAA гена UGT1A1. Несмотря на это, возможно возникновение более ста иных мутаций, которые стали причиной возникновения заболевания. Такие проявления происходят изредка, но возможно.
Вчера было все в норме. Почему билирубин повысился именно сейчас?
Организм человека выводит различные вещества из «зоны комфорта», что приводит к увеличению объема билирубина. Этому может поспособствовать голодовка (диета), чрезмерное обезвоживание, наличие острых заболеваний или обострение хронических недугов, стрессовое состояние, цикл менструации у женщины и прочее.
Чувствую себя плохо. Доктор сказал, что билирубин здесь не при чем. Так ли это?
Не стоит паниковать по каждому случаю. Возможны жалобы на наличие недомогания, усталости, тошноты и беспокойства, но итоговые исследования не укажут на связь с симптомом Жильбера. Уровень билирубина не высокий. Причин для беспокойства нет.
Список литературы
Клинический протокол диагностики и лечения синдрома Жильбера. Министерство здравоохранения РК. 2014.
Люди X. Синдром ломкой X-хромосомы
Вы встречали в своей жизни человека с синдромом Дауна? Скорее всего, да. А с синдромом ломкой Х-хромосомы? Скорее всего, тоже да, просто вы об этом не знали. По статистике, носителями этого синдрома являются примерно 1 из 4 000 мужчин и 1 из 8 000 женщин. А еще это вторая по распространенности, после синдрома Дауна, генетическая причина умственной отсталости и самая частая моногенетическая причина аутизма.
Кажется, мы уже хорошо знаем, что человек с аутизмом — это не слегка нелепый «человек дождя» или гениальный программист, который просто не понимает шуток. И, кажется, мы довольно быстро привыкли к слову «аутизм», а некоторые даже знакомы с термином РАС. Но что подразумевает этот самый аутистический спектр, что он в себя включает? А из-за чего бывает аутизм? Ученый, который ответил бы на этот вопрос, получил бы все нобелевские премии и все почетные ордена. Но такого ученого нет и не будет, потому что аутизм — это не одна конкретная болезнь с одной конкретной причиной, это много причин, часто генетических.
А вот синдром ломкой Х-хромосомы — очень конкретное состояние и очень конкретный диагноз, который можно подтвердить одним конкретным анализом. И в некоторых странах, например, существует диагностический стандарт, по которому всем мальчикам с аутизмом делают этот анализ. А еще бывает, что анализ на носительство премутации (она может привести к полной мутации) делают женщинам, которым сложно забеременеть или у которых наступила ранняя менопауза.
Почему же мы так плохо знакомы с этим состоянием? Может быть, потому что этот синдром не проявляется таким характерным фенотипом, как синдром Дауна? А может быть, потому что аутизм и умственная отсталость кажутся более серьезными проблемами, чем другие клинические проявления синдрома, которых немало и за которыми тоже стоит следить? Зато синдром ломкой Х-хромосомы очень любят ученые, занимающиеся генной терапией, так как поломка, вызывающая эту мутацию, представляет собой удобную мишень для испытаний и экспериментов, которые проводят в поисках лечения состояний, вызывающих аутизм.
Наследование и развитие синдрома зависит от пола, так как ген FMR1, вызывающий это заболевание, находится на Х-хромосоме. У мужчин только одна Х-хромосома, которую они получают от матери. Поэтому в случае, если мать является носительницей синдрома и передала сыну «ломкую» хромосому, у него проявится заболевание. У женщин две Х-хромосомы, однако активно работает только одна из них. Поэтому в случае инактивации этой «ломкой» Х-хромосомы заболевание может не проявляться клинически или приводить к развитию заболевания только в некотором проценте случаев. Мужчина с ломкой Х-хромосомой может передать ее всем дочерям, но ни одному из сыновей. Женщина может передать ее как сыновьям, так и дочерям с равной вероятностью.
Поведенческие характеристики могут включать (а могут и не включать!) синдром дефицита внимания (СДВ), синдром дефицита внимания и гиперактивности (СДВГ), аутизм и аутистическое поведение, социальное беспокойство, привычку хлопать в ладоши, трудно устанавливаемый зрительный контакт, сенсорные расстройства и повышенный риск агрессии.
Для большинства мальчиков с синдромом ломкой X-хромосомы характерны значительные интеллектуальные нарушения — от умеренного отставания в обучении до более серьезных проблем.
К физическим особенностям мужчин постпубертатного возраста можно отнести большие уши, вытянутое лицо, чрезмерно растяжимую кожу и увеличение размера яичек (так называемый макроорхидизм). Вовлечение в патологический процесс соединительной ткани приводит к таким нарушениям, как отиты, плоскостопие, готическое небо, патологическая гибкость пальцев и гиперподвижность суставов. Ни у одного человека не будут представлены сразу все признаки, а некоторые симптомы, такие как удлиненное лицо и макроорхидизм, чаще встречаются у мужчин после полового созревания.
Бывает так, что у человека с синдромом ломкой Х-хромосомы нет классических признаков аутизма, а есть отдельные проявления типа фиксированных интересов или сложностей с нарушениями рутины. Интересно, что люди с синдромом ломкой Х-хромосомы очень общительны и дружелюбны, имеют отличные навыки подражания, обладают хорошей зрительной и долговременной памятью, любят помогать другим; это люди с прекрасным чувством юмора. Их высокая чувствительность помогает и мешает им одновременно, так как, с одной стороны, они очень эмпатичны, с другой стороны, очень чутко реагируют на плохое настроение окружающих или конфликтную ситуацию, даже потенциальную или не имеющую к ним никакого отношения.
Симптомы, наблюдаемые у мужчин, могут также встречаться и у женщин. Однако женщины часто имеют не такие выраженные интеллектуальные нарушения, а также умеренные поведенческие и физические отклонения.
Но все же примерно треть женщин с диагностированным синдромом значительно отстает в умственном развитии. У остальных могут встречаться легкие или умеренные нарушения обучаемости, эмоциональные/психические отклонения, общее беспокойство и/или социальная тревожность. Небольшой процент женщин-носительниц полной мутации гена FMR1, которая обусловливает развитие синдрома, не имеет четких признаков заболевания — интеллектуальных, поведенческих или физических нарушений. У этих женщин синдром часто выявляется только после того, как заболевание было диагностировано у другого члена семьи.
Зачем вообще ставить диагноз, если специфического лечения нет? Для того, чтобы предупредить остальных членов семьи, для того, чтобы иметь возможность планировать беременность и для того, чтобы знать, что у женщины в принципе могут быть сложности с планированием беременности, если у нее есть премутационный статус. Статус премутации сам по себе — состояние, которое также может сопровождаться определенными нарушениями. В пожилом возрасте у мужчин может развиться синдром тремор/атаксии (FXTAS) — тремор, шаткая походка, может страдать речь. Женщины могут столкнуться с так называемой первичной недостаточностью яичников (FXPOI) (снижение овариального резерва и наступление менопаузы до 40 лет). Среди носительниц премутации примерно у четверти развивается это состояние. Оно влияет не только на общие репродуктивные возможности, но и часто оказывается причиной скудного ответа яичников на стимуляцию. Хороший невролог и гинеколог обязательно должны знать о том, что у их пациента есть этот диагноз.
Если нет специфического лечения, это не значит, что нет специфических методов и стандартов поведенческого или педагогического вмешательства, которые помогут и родителям, и представителям помогающих профессий, и учителям, и, конечно, самим носителям синдрома.
Найти больше информации о синдроме и познакомиться с сообществом родителей и специалистов можно здесь:
Григорьева Татьяна Витальевна
член Ассоциации поддержки людей с синдромом ломкой Х-хромосомы
Болезнь стальных волос: есть ли шанс у людей с синдромом Менкеса?

Болезнь Менкеса относится к наследственным генетическим заболеваниям и встречается с частотой 1 случай на 40-350 тысяч новорожденных мальчиков. Нехватка меди, с которой связано развитие данной патологии, очень быстро сказывается на состоянии ребёнка. Без лечения такие пациенты заметно отстают в развитии уже с первых месяцев и умирают к 2-3 годам. Как и все генетические болезни, синдром Менкеса очень сложно поддается терапии. Но ученые с каждым годом все больше совершенствуют существующие методы лечения и разрабатывают новые. О них и расскажет MedAboutMe.
Что такое болезнь Менкеса: причины и симптомы

Болезнь Менкеса — генетическое заболевание, которое проявляется только у мальчиков. Аномалия при этом наследуется от матери, у которой в ДНК присутствует патологический ген ATP7A. Несмотря на то что болезнь передается именно от женщин, сами они могут быть только носителями патологии.
Проявляется синдром Менкеса в плохом усваивании меди организмом. В результате ребёнок еще в утробе начинает страдать от нехватки этого микроэлемента. Дети часто рождаются недоношенными.
Симптомы болезни начинают проявляться в первые месяцы жизни. Дефицит меди выражается в таких признаках:
- Запутанные, ломкие, веретенообразные волосы. Этот симптом настолько характерен, что синдром часто называют болезнью стальных или курчавых волос. Исследование волосинок под микроскопом является одним из способов диагностики заболевания.
- Задержка физического развития.
- Проблемы с кормлением.
- Постоянное расстройство пищеварения — хроническая диарея.
- Судороги.
- Белая дряблая кожа (медь влияет на выработку коллагена), при этом новорожденные часто отличаются слишком полными румяными щеками.
- Отсутствие эмоций (вследствие нарушения продукции дофамина).
- Остеопороз, другие проблемы с костями.
Болезнь прогрессирует, и с возрастом симптомы становятся более выраженными. Нехватка меди сказывается на работе головного мозга, поэтому родители могут отмечать у ребёнка задержку психического развития. У детей с синдромом Менкеса часто возникают воспалительные инфекционные болезни, причиной смерти может стать сепсис или пневмония.
Несмотря на неблагоприятные прогнозы, современное лечение все же дает таким детям шанс на жизнь. Известен случай пациента с болезнью Менкеса, который дожил до совершеннолетия. В 2016 году Блейн Гренон (Blaine Grenon) отметил 21-летие. Молодой человек не только справляется с болезнью, но даже обучается в коллеже (Community College of Rhode Island). Несмотря на то что этот случай уникален, история пациента Гренона дает шанс и другим больным синдромом Менкеса.
Диагностика болезни
Ранняя диагностика болезни Менкеса — важная предпосылка ее успешного лечения, ведь заболевание прогрессирует, и каждый месяц пропущенной реабилитации существенно ухудшает состояние больного. Заподозрить генетические нарушения могут еще неонатологи при осмотре новорожденного. Однако точный диагноз можно поставить только на основании анализов и обследований. Для этого ребёнку назначается:
- Анализ крови и мочи на содержание меди.
- Рентген скелета (выявляет патологию в костях).
- МРТ/КТ головного мозга (обнаруживаются атрофии, в том числе мозжечка, фокальные очаги некроза).
- Микроскопическое исследование волос.
Диагностика болезни Менкеса всегда комплексная, в частности, дополнительно может проводиться биопсия печени для выявления уровня меди или другие исследования.
Важную роль регулярные обследования играют и для оценки результатов лечения у ранее диагностированного больного. Так, в 2014 году Крис Чанг (Chris Chang) и группа исследователей Калифорнийского университета в Беркли разработали несколько типов флуоресцентных зондов, помогающих определить количество меди в головном мозге. «Ранее отсутствовал мониторинг изменений содержания меди в организме. Поэтому врачам было трудно определить, как количество микроэлемента отражается на разных этапах болезни», — отмечает исследователь. С помощью новых зондов, в частности Copper Fluor-3 (CF 3), можно держать под контролем любые изменения количества меди. Зонды разработаны для широкого спектра обследований, но, безусловно, для пациентов с болезнью Менкеса являются очень важными.
Лекарства: заместительная терапия медью
Основой лечения болезни Менкеса в современной медицине является заместительная терапия медьсодержащими препаратами. Одним из распространенных вариантов является медьсодержащий гистидин (незаменимая аминокислота), который пациент должен принимать ежедневно на протяжении всей жизни. При таком лечении уровень меди стабилизируется уже через 2-3 недели. Но главная проблема подобных препаратов — при длительном применении медь может накапливаться в почках, что сказывается на их работе. При этом препарат считается наиболее действенным, и сегодня продолжаются исследования, связанные с его использованием. Так, в 2017 году Cyprium Therapeutics, дочерней компанией Fortress Biotech, был предложен препарат CUTX-101 — подкожная инъекция гистидината меди. Согласно исследованиям, лекарство улучшает показатели выживаемости среди пациентов с синдромом Менкеса. Сейчас компания ведет переговоры с FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) о производстве препарата и его использовании для лечения людей с этим генетическим заболеванием.
В 2014 году ученые центра биотехнологий RIKEN в Кобе (Япония) совместно с педиатрами из Городского университета Осаки провели исследования с помощью позитронно-эмиссионной томографии (ПЭТ). Использование ПЭТ позволило оценить распределение меди в организме после введения различных препаратов. В результате исследование показало, что комбинация инъекций меди и дисульфирама или D-пеницилламина способствует правильному распределению микроэлемента — большая его часть оказывается в мозге, а в почках он не накапливается. Исследование проводилось на мышах — одной группе вводился просто изотоп меди, другой ставили комбинированные инъекции с дисульфирамом. У животных из второй группы наблюдалось лучшее распределение микроэлемента по организму. Использование ПЭТ-диагностики для больных синдромом Менкеса позволит в дальнейшем эффективнее оценивать действие выписываемых лекарств.
Генная терапия
Поскольку синдром Менкеса — это генетическое заболевание, любые лекарства являются лишь временной мерой и привести к полному выздоровлению не могут. Для того чтобы добиться полноценных результатов, нужно вносить изменения в ДНК — убирать неправильно работающие гены. Поэтому наиболее перспективным направлением сегодня считается генная терапия. На данном этапе все новые технологии проходят испытания и пока не применяются для лечения реальных пациентов.
Одной из самых популярных сегодня технологий, которая в перспективе позволит лечить наследственные заболевания, является система CRISPR-Cas. По своей сути технология подобна «ножницам», которые будут внедряться в клетку, искать аномальный участок и удалять его. Молекула ДНК после такой процедуры будет восстанавливаться, дублируя оставшуюся здоровую часть цепи, а значит, избавляться от патологий, которые приводят к заболеванию. С помощью CRISPR-Cas теоретически можно будет лечить многие болезни, причиной которых является один аномальный ген. Технология пока очень несовершенна, но перспективы у нее достаточно обнадеживающие.
Побороть болезнь Менкеса с помощью генной инженерии предложила и компания Cyprium Therapeutics. Технология AAV-ATP7A предполагает создание здоровых генов ATP7A и внедрение их в организм больного. Клинические исследования метода назначены на 2018 год.
Важную роль в генной терапии болезни Менкеса сыграло и исследование 2015 года, которое провели ученые Университета Миссури. Их задачей было выяснить, во всех ли клетках организма ген ATP7A играет важную роль. Опыты на мышах показали, что в некоторых случаях эта патология не сильно отражается на состоянии здоровья. Например, если мутация присутствовала только в клетках центральной нервной системы, это не существенно отражалось на здоровье. У мышей с такой аномалией болезнь Менкеса проявлялась лишь незначительными неврологическими симптомами. «Если мы будем знать, какие органы или ткани больше всего ответственны за усвоение меди, в лечении мы можем сосредоточиться именно на этих областях», — отметил ведущий исследователь Майкл Петрис.
Болезнь или синдром Шёгрена
В этом месяце жителям районов Савеловский, Беговой, Аэропорт, Хорошевский предоставляется скидка 5% на ВСЕ медицински.
Скидки для друзей из социальных сетей!
Эта акция - для наших друзей в "Одноклассниках", "ВКонтакте", "Яндекс.Дзене", YouTube и Telegram! Если вы являетесь другом или подписчиком стр.
Гуляев Сергей Викторович
Врач-ревматолог, терапевт, нефролог
Кандидат медицинских наук
Мы в Telegram и "Одноклассниках"
Болезнь или синдром Шёгрена - системное аутоиммунное заболевание, известное также как «сухой синдром». Болезнь Шёгрена получила свое название в честь шведского офтальмолога Генриха Съогрена. В 1929 году он обследовал и лечил пациента, которого сильно тревожили сухость в глазах, во рту и боль в суставах.
У женщин болезнь Шёгрена встречается намного чаще, чем у мужчин (в 9 из 10 случаев), причем, как правило, это касается женщин после менопаузы. Однако в целом заболевание поражает людей любого пола и возраста. Общемировой статистики нет, но в развитых странах, включая Россию, заболеваемость оценивается в миллионах, болезнь Шёгрена - одно из самых распространённых ревматических заболеваний.
При болезни Шёгрена иммунитет человека воспринимает клетки собственного организма как чужие и начинает их медленно и планомерно уничтожать. Клетки иммунной системы попадают в ткани желез внешней секреции (слезные, слюнные, бартолиниевы железы влагалища), поражают их, и те начинают выделять меньше соответствующего секрета (слюны, слез и т.д.).
Кроме того, болезнь нередко затрагивает и иные органы, провоцируя артралгии, боли в суставах, мышцах (полимиозит), одышку и т.д.
Есть также синдром Шёгрена (вторичное воспаление слюнных и слезных желез), который сопутствует ревматоидному артриту, диффузным болезням соединительной ткани, заболеваниям желчевыводящей системы и другим аутоиммунным заболеваниям.

Диагностика болезни Шёгрена


Важно обратиться к врачу при первых симптомах, так как «запущенная» болезнь Шёгрена может принять неблагоприятный характер и затронуть жизненно важные органы, что нередко приводит к осложнениям, в редких случаях - к летальному исходу.
Причины развития
Одним из главных факторов, «запускающих» болезнь, является аутоиммунный сбой. При данном нарушении иммунная система начинает уничтожать клетки желез внешней секреции человека. Почему это происходит? Этот механизм при болезни Шёгрена еще нуждается в уточнении.
Другим фактором появления заболевания является генетическая предрасположенность. Иногда, если эта болезнь есть у матери, то она может быть выявлена и у дочери. Изменения в гормональном фоне женщины тоже могут спровоцировать болезнь.
Синдром Шёгрена обычно развивается на фоне других системных заболеваний (напр., при ревматоидном артрите и системной красной волчанке).
Клиническая картина болезни Шёгрена
Все симптомы болезни Шёгрена можно условно разделить на железистые и внежелезистые.
Железистые симптомы болезни Шёгрена
Железистые симптомы болезни проявляются в снижении выработки секретов желез.
Одним из основных признаков болезни Шёгрена является воспаление глаз, связанное с уменьшением секреции глазной жидкости. Больных беспокоит чувство дискомфорта: жжение, царапанье, «песок» в глазах. Вместе с этим люди часто ощущают отек век, покраснение, скопление в углах глаз белой вязкой жидкости. На следующем этапе заболевания пациенты начинают жаловаться на светобоязнь, ухудшение остроты зрения.
Второй постоянный признак болезни Шёгрена - воспаление слюнных желез, которое переходит в хроническую форму. Больной жалуется на сухость во рту и увеличение слюнных желез. В начале болезни отмечается небольшая или непостоянная сухость во рту, которая появляется только в результате волнения или физической нагрузки. Затем сухость во рту становится постоянной, слизистая оболочка и язык чрезмерно сохнут, приобретают ярко розовый цвет и часто воспаляются, быстро прогрессирует зубной кариес.
Иногда до появления этих признаков у больного может появиться «беспричинное» увеличение лимфатических узлов.
Поздняя стадия болезни характеризуется сильной сухостью во рту, человеку становится очень сложно разговаривать, проглатывать твердую пищу, не запивая ее водой. На губах появляются трещины. Может появиться хронический атрофический гастрит с недостаточностью секреции, которая сопровождается отрыжкой, тошнотой, снижением аппетита. У каждого третьего больного на поздней стадии отмечается увеличение околоушных желез.
Наблюдается поражение желчных путей (холецистит), печени (гепатит), поджелудочной железы (панкреатит).
На поздней стадии заболевания становится очень сухой носоглотка, в носу образуются сухие корочки, может развиться отит и снижение слуха. Из-за сухости в гортани появляется осиплость голоса.
Появляются вторичные инфекции: часто рецидивирующие синуситы, трахеобронхиты, пневмонии. У каждой третьей больной наблюдается воспаление половых органов. Слизистая оболочка красная, воспаленная.
Синдром Марфана

Синдром Марфана — наследственное заболевание, которое проявляется системным поражением соединительной ткани в организме человека. В результате болезни происходят нарушения строения скелета и кожи, работы глаз, сердечно-сосудистой, дыхательной и других систем организма. Эту генетическую мутацию нельзя предотвратить или вылечить, но правильно подобранное лечение способно продлить пациентам жизнь и предупредить развитие опасных осложнений.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
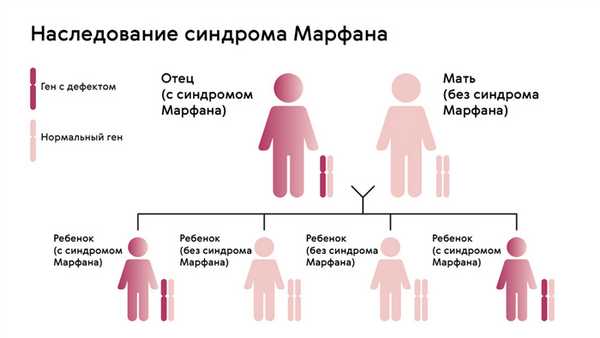
Рисунок. 1. Схема наследования синдрома Марфана. Источник: МедПортал
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на 5-10 тысяч.
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно. Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25-50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
Симптомы синдрома Марфана
Проявления генетического дефекта могут быть выражены в разной степени: от легкого изменения строения соединительной ткани до тяжелых нарушений жизненно важных функций организма. Иными словами, внешние признаки аномалии у разных пациентов могут значительно отличаться, несмотря на одинаковый генетический дефект.
Классической триадой синдрома Марфана считаются: скелетные нарушения, смещение хрусталика и расслоение аорты (рис. 2). Также системное поражение соединительной ткани у пациентов становится причиной развития нарушений работы практически всех органов и систем организма.
Костно-мышечная система
Выраженность симптомов поражения опорно-двигательного аппарата зависит от тяжести случая и особенностей организма пациента.
Для людей с синдромом Марфана характерен чрезвычайно высокий рост: обычно дети «перерастают» всех членов семьи. При этом часто, особенно в детском возрасте, привлекает внимание нестандартная длина рук: их размах оказывается больше, чем длина тела.
Яркий симптом болезни — патологически удлиненные и тонкие пальцы, так называемые «пальцы паука» (арахнодактилия) (рис. 3).
Проверить наличие симптома можно с помощью теста большого пальца кисти — у пациентов с арахнодактилией часть большого пальца (дистальная фаланга) выступает за край сжатой в кулак ладони (рис. 4).
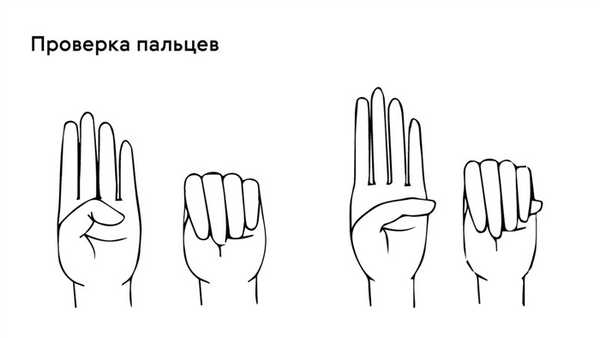
Рисунок 4. Проверка на арахнодактилию. Источник
Лицо людей с синдромом Марфана обычно вытянутое и худое. Этому способствует высокое положение свода верхнего неба, удлиненный череп и патологическая худоба.
Также для таких пациентов характерны деформации грудной клетки, которые могут быть в двух вариантах: смещение грудины внутрь (воронкообразная грудь) или наружу (килевидная грудь, рис. 5).
Осанка пациентов с синдромом Марфана в большинстве случаев нарушена. Чаще всего определяются различные степени выраженности сколиоза (отклонение позвоночного столба в сторону) или кифоза (формирование «горба»).
Кроме того, пациенты с FBN1 мутацией часто страдают от:
- плоскостопия;
- повышенной подвижности всех суставов;
- слабости связочного аппарата.
У пациентов с синдромом Марфана часто плохо развиты мышечные структуры и практически нет подкожно-жирового слоя. Движения пациентов с этой патологией неловкие, они часто получают различные травмы.
Высокий темп роста и нарушения выработки белков соединительной ткани определяют патологии кожи у людей с мутацией гена FBN1. Клинически это проявляется в виде повышенной растяжимости кожных структур с образованием светлых полос — «растяжек» (стрий).
Зрение
Дефекты гена FBN1 определяют склонность к патологиям зрительной системы. Чаще всего повреждения глаз у пациентов с синдромом Марфана включают в себя:
- выраженную близорукость;
- подвывих или изменение положения хрусталика;
- высокий риск внезапной отслойки сетчатки глаза.
Кроме того, у таких пациентов гораздо раньше может развиться катаракта или глаукома: те патологии органа зрения, которые считаются возрастными у здоровых людей.
Органы дыхания
В легких пациентов с синдромом Марфана может патологически разрастаться соединительная ткань. Это приводит к формированию сужения бронхов и легочного фиброза. Нередко на фоне генетической мутации развивается бронхиальная астма или хроническое обструктивное заболевание легких. Генетическая аномалия также определяет возможность развития спонтанного пневмоторакса — неотложной ситуации, в которой в полость вокруг легких попадает воздух, и легкое резко уменьшается в размерах («спадается»).
Желудочно-кишечный тракт
Процессы пищеварения у людей с FBN1 мутацией меняются: нарушается моторика кишечника, появляются патологии желчного пузыря, часто развиваются гастриты, язвенные дефекты, дисбиоз.
Почечный аппарат
У пациентов с синдромом Марфана чаще находят аномалии почек: опущение органов, расширение почечных лоханок, патологическую подвижность почек.
Нервная система и психическая сфера
Хотя в большинстве случаев у пациентов с синдромом Марфана не происходит нарушений работы мозговых структур, некоторые патологические изменения нервной системы могут присутствовать. Например, расширение соединительнотканной капсулы, которая окружает спинной мозг, может приводить к нарушениям движений в нижних конечностях, работы мочевого пузыря и кишечника. Для таких пациентов характерно развитие синдрома хронической усталости — астения, склонность к депрессии. Интеллектуальная деятельность в большинстве случаев не нарушена, даже наоборот: среди пациентов с синдромом Марфана есть люди с интеллектом значительно выше среднего.
Сердечно-сосудистая система
Кардиологи выявляют нарушения ритма сердца у людей с синдромом Марфана. У пациентов с этой патологией часто нарушается структура аортального клапана — соединительнотканной перегородки, которая предупреждает обратный ток крови из аорты в сердце. Это приводит к развитию порока сердца — аортальной недостаточности. Также могут развиваться другие пороки сердца, например, пролапс или недостаточность митрального клапана, а на пораженных участках часто развивается инфекционно-воспалительный процесс — бактериальный эндокардит.
Самую большую опасность представляют патологические изменения в главном сосуде организма — аорте. У 65-100% людей с синдромом Марфана есть большой риск поражения луковицы (наиболее близкая к сердцу часть аорты) и восходящей дуги этой артерии — тех частей, которые непосредственно выходят из сердца. Поскольку внутренний слой стенки сосудов также содержит волокна соединительной ткани, они склонны к износу, а давление крови в аорте выше, чем в других участках сосудистого русла. Это приводит к тому, что сосуд постепенно расширяется, и может произойти патологическое скопление крови между сосудистыми стенками с формированием мешковидного выпячивания (аневризмы) или спонтанный разрыв артерии.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка. При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7-18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка. Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны. Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития.
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает 70 лет. Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.
Заключение
Конечно, жизнь с этой генетической мутацией становится сложнее, но при правильном подходе к собственному здоровью и своевременному обследованию у врачей пациентам с синдромом Марфана удается компенсировать все проявления заболевания и не допустить развития фатальных осложнений.
Активисты с синдромом Марфана создают тематические сообщества по всему миру: мощная поддержка людей с такой же генетической аномалией позволяет пациентам не чувствовать себя одинокими.
Читайте также:
- Головная боль и давление при предменструальном синдроме. ЭКГ при ПМС и вегетативно-дизовариальная миокардиодистрофия
- Диагностика, дифференциация, лечение и прогноз хромофобной аденомы гипофиза
- Денервационная боль. Лечение хронической боли. Принципы лечения хронической боли.
- УЗИ, МРТ при клапане задней уретры у плода
- Нечеткость контуров органов брюшной полости. Закрытые травмы живота