Синдром Олбрайта-Хадорна (Albright-Hadorn) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 22.01.2026
синдром Олбрайта
псевдогипопаратиреоз
НЗЧ, характеризующееся изменениями скелета и нарушением фосфорно-кальциевого обмена, что приводит к задержке роста, укорочению пальцев, образованию подкожных кальцификатов, отмечаются судороги, умственная отсталость и др.; передается по аутосомно-рецессивному типу; связывается с мутациями гена GNAS1, кодирующего гуанин-связывающий белок и локализованного на хромосоме 20.
[Арефьев В.А., Лисовенко Л.А. Англо-русский толковый словарь генетических терминов 1995 407с.]
Тематики
Синонимы
2 синдром Олбрайта
3 синдром Олбрайта
4 синдром
синдром верхней части плечевого сплетения — невр. Duchenne-Erb palsy, Duchenne-Erb syndrome, Duchenne-Erb paralysis
синдром висцеромегалии и офтальмоцеле — Beckwith-Wiedemann [visceromegaly and ophthalmocele] syndrome
синдром гипоплазии бедра и необычного лица — Franz's [femoral hypoplasia-unusual facies] syndrome
синдром диссеминированной внутрисосудистой коагуляции — disseminated intravascular coagulation, DIC
синдром идиопатической гиперкальциемии — idiopathic hypercalcemia [Williams] syndrome
синдром передних рогов спинного мозга — невр. anterior horns of spinal cord syndrome
синдром повышенной раздражимости толстой кишки — irritable colon [irritable bowel] syndrome
синдром половинного поражения спинного мозга — hemisection of spinal cord [Brown-Séquard] syndrome
синдром приобретённого иммунодефицита — acquired immune deficiency syndrome, AIDS
синдром прогрессирующего супрануклеарного паралича — невр. progressive supranuclear palsy [Steele-Richardson-Olszewski] syndrome
латеральный синдром продолговатого мозга — невр. lateral aspect of medulla [Wallenberg-Zacharchenko] syndrome
5 Олбрайта наследственная остеодистрофия
6 синдром Клайнфелтера-Рейфенштейна - Олбрайта
Medicine: Klinefelter's syndrome (сочетание хромосомного нарушения XXV, первичного гипогонадизма, олиго- и азооспермии)
7 синдром Лайтвуда-Олбрайта
8 синдром Мак-Кьюна-Олбрайта
9 синдром Мартина - Олбрайта
10 синдром Мартина-Олбрайта
11 синдром Форбса - Олбрайта
12 синдром Форбса-Олбрайта
13 синдром Фулера-Олбрайта
14 синдром Лайтвуда-Олбрайта
15 синдром Мартина-Олбрайта
См. также в других словарях:
синдром Олбрайта — синдром Олбрайта. См. псевдогипопаратиреоз. (Источник: «Англо русский толковый словарь генетических терминов». Арефьев В.А., Лисовенко Л.А., Москва: Изд во ВНИРО, 1995 г.) … Молекулярная биология и генетика. Толковый словарь.
синдром Олбрайта — псевдогипопаратиреоз НЗЧ, характеризующееся изменениями скелета и нарушением фосфорно кальциевого обмена, что приводит к задержке роста, укорочению пальцев, образованию подкожных кальцификатов, отмечаются судороги, умственная отсталость и др.;… … Справочник технического переводчика
Синдром Олбрайта-Мак-Кьюна-Штернберга — Проявляется в детском возрасте. Этиология не известна. Проявляется нарушением функций шишковидной железы и других структур промежуточного мозга, что ведет к избытку продукции гонадотропинов и к преждевременному половому развитию. Вместе с тем… … Энциклопедический словарь по психологии и педагогике
Синдром Олбрайта-Хадорна — Первичное нарушение калиевого обмена с периодическими гипокалиемическими параличами мышц. Проявляется пароксизмами общей слабости, болями в костях, спине, остеопорозом костей, гипо- или арефлексией, парестезиями, отеками, олигурией и… … Энциклопедический словарь по психологии и педагогике
Олбрайта болезнь — (F. Albright, род. в 1900 г., амер. врач.; син: Олбрайта синдром, Олбрайта Мак Кьюна Штернберга болезнь) болезнь неясной этиологии, проявляющаяся преждевременным половым созреванием, множественной фиброзной остеодисплазией и гиперпигментацией… … Большой медицинский словарь
Олбрайта болезнь — I Олбрайта болезнь (F. Albright, амер. врач, родился в 1900 г.) см. Псевдогипопаратиреоз. II Олбрайта болезнь (F. Albright, р. 1900 г., американский врач.; син: Олбрайта синдром, Олбрайта Мак Кьюна Штернберга болезнь) болезнь нежной этиологии,… … Медицинская энциклопедия
Синдром Клайнфельтера — Кариотип … Википедия
Синдром поликистозных яичников — Поликистозный яичник: ультразвуковое изображение МКБ 10 E … Википедия
Синдром Ди Джоржи — Синдром Ди Джорджа … Википедия
Синдром Шихана — Синдром Шихана(шэегана) МКБ 10 E23.023.0 МКБ 9 253.2253.2 DiseasesDB … Википедия
Синдром Олбрайта

Синдром Мак-Кьюна-Олбрайта является гетерогенным заболеванием, которое независимо друг от друга описали американский педиатр D. J. McCune и эндокринолог F. Albright в 1936-1937 гг. Заболевание редкое, врожденное, но не наследственное, сопровождается метаболическими нарушениями, признаками преждевременного полового развития и деформаций скелета. Первые клинические проявления отмечаются в раннем детстве или подростковом возрасте в виде нарушений развития, полиоссальной фиброзной дисплазии костей, кожной пигментации по типу «кофе с молоком» и эндокринных аномалий, включая тиреотоксикоз, синдром Кушинга, акромегалию.
Приблизительно в 10 раз чаще диагностируют синдром Олбрайта Мак-Кьюна у девочек, чем у мальчиков. Википедия отмечает, что частота встречаемости в человеческой популяции — 1 на 100 тыс. или на миллион особ.
Патогенез
В результате постзиготной мутации гена GNAS1, участвующего в клеточном сигнализировании белков GS-α , обеспечивающих сопряжение рецепторов лютеинизирующих и фоликолостимулирующих гормонов с молекулами аденилатциклазы в цитологических структурах яичников. Постоянное влияние мутантного белка активизирует аденилатциклазу и повышение внутриклеточного цАМФ, а также стимулирует усиление секреции стероидных женских половых гормонов фолликулярными эстрогенпродуцирующими кистами, вызывающими преждевременное половое развитие даже при отсутствии гонадотропинов. Такая же картина наблюдается и «автономное производство» наблюдается с гормонами щитовидной железы, кортизолом и гормонами роста. Прогрессирование патологии вызвано образованием клеток, уже содержащих мутантные белки.
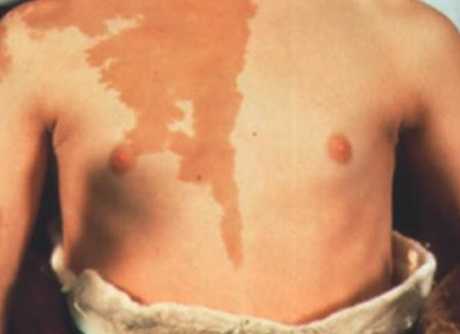
Пигментация при синдроме Олбрайта
Кроме того, рост уровня цАМФ приводит к увеличению продукции меланина и образованию лентигиозных пятен, очаговой гиперпигментации на фоне нормальных значений АКТГ и меланотропина.
Фиброзно-кистозная дисплазия в результате усиленной кальцинации под действием эстрогенов осуществляется путем замещения нормальных костных структур и образованных кальцинатов фиброзными массами, при этом нарушается равновесие процессов резорбции и регенерации костей, пролиферации мезенхимальных стромальных клеток. Наблюдается формирование очагов деструкций и псевдоопухолей.
Классификация
Болезнь Олбрайта бывает разной, в зависимости от тяжести течения различают:
- скрытое заболевание - без каких-либо признаков костной или эндокринной патологии, а также без симптоматики необычной пигментации;
- неполные формы синдрома, например, в процесс патогенеза вовлечения костная ткань, возможны эндокринные изменения и псевдогипопаратиреоз, но нет кожных проявлений;
- псевдогипопаратиреозили наследственная остеодистрофия Олбрайта — заболевание, которое также поражает костные ткани, но механизм и этиология отличаются — патология вызвана нарушением резистентности к паратгормону, метаболизма кальция и фосфора; псевдогипопаратиреоз также проявляется в детском возрасте и выражается в виде низкого роста, круглого лица, умственной отсталости, ожирения, гипотиреоза, гипогонадизма, сахарного диабета и пр.
Причины
Синдром Олбрайта Маккьюна возникает в результате генетических нарушений на ранних этапах эмбриогенеза. До сих пор не установлен тип наследования, все описанные случаи были спорадические.
Симптомы
Клиническая картина заболевания проявляется в виде целого симпатокомплекса, состоящего из:
- преждевременного гонадотропиннезависимого полового развития, развивающегося позже и медленнее нежели при других формах ранней половозрелости;
- пятнистой пигментации кожи - лентиго, пятна с четкими границами светло-коричневого цвета обычно есть от рождения на груди, спине пояснице, бедрах и в местах костных деформаций;
- эндокринных расстройствах, вызывающих тахикардию, необъяснимую утрату веса, глазные симптомы гипертиреоидизма;
- полиоссальной фиброзной остеодисплазии, мышечной слабости и остеопороза, вовлекающей в процесс чаще длинные кости, нарушения проявляются в виде изуродованных черт лица, более вытянутом телосложении, искривленных и асимметричных, укороченных запястьях, что затрудняет движения во время ходьбы, вызывает хромоту, сильные боли и повышает вероятность патологических переломов.

Первые признаки преждевременной половой зрелости начинаются с маточных кровотечений, не имеющих цикличного характера и вызваны кратковременными повышениями концентраций эстрогенов и гонадотропных гормонов в кровяном русле. Такие колебания провоцируют образования кист в яичниках. У детей в возрасте 5-10 лет наблюдается ранний рост груди и гинекомастия. Её нежная этиология не приводит к формированию вторичных половых признаков — Синдром Мак-Кьюна-Олбрайта-Брайцева не вызывает раннего полового оволосения или дифференцировки фигуры. Истинное половое созревание возможно после окончания пубертатного периода.
Анализы и диагностика
При подозрении на синдром Мак Кьюна Олбрайт проводят:
- рентгенологические исследования, которые выявляют псевдокисты, сегментарное поражение любых отделов скелета - чаще нижних конечностей, в более редких случаях — верхнего плечевого пояса и черепа;
- УЗИ брюшной полости позволяет обнаружить кисты яичников;
- лабораторные исследования для выявления повышенного уровня щелочной фосфатазы в кровотоке, сниженного количества гонадотропинов - в анализах мочи и повышенной экскреции лютеинизирующего гормона, 17-гидрокси и 17-оксикортикостероидов.
Лечение
Лечение назначают симптоматическое, оно имеет свои сложности в зависимости от индивидуальных особенностей и клинической картины. Аналоги гонадолиберина не оказывают терапевтического эффекта, так как процесс секреции эстрогенов спровоцирован не избытком гонадотропинов. Для купирования процесса гонадального стероидогенеза у особ мужского пола эффективным может оказываться кетоконазол и андрокур. При лечении медроксипрогестерона ацетатом возможно развитие гипокальциемии, поэтому, если процесс развития ранней половозрелости сопровождается тяжёлыми множественными поражениями костных тканей, то терапию необходимо назначать с осторожностью.
Для угнетения гиперэстрогенизиции и удлинения скелета могут быть назначены препараты тестостерона. Чтобы противодействовать дисплазии костей следует вводить:
- бисфосфонаты;
- ингибиторы антагонистов остеокластов.
Больным необходимо регулярно, каждые 3 мес. посещать эндокринолога для коррекции проблем с щитовидной и другими железами.
ОЛБРАЙТА СИНДРОМ
наследственная остеодистрофия Олбрайта, по имени американского врача эндокринолога F. Albright, 1900-1969; синоним - псевдогипопаратиреоз) - фиброзная остеодисплазия нескольких костей скелета (порок развития костной ткани с частичным замещением ее хрящевой) и очаговая пигментация кожи в сочетании с эндокринными расстройствами. Основные проявления: симптомы гипопаратиреоза, связанные с невосприимчивостью тканей к паратгормону; изменения скелета. Диагностируют заболевание чаще между 5-м и 10-м годами жизни на основании низкого роста, диспропорции скелета с преобладанием укорочения нижних конечностей, короткой шеи, круглого лица, короткопалости с преимущественным укорочением I, III, IV, V метакарпальных и метатарзальных костей, из-за чего при сжатии руки в кулак отсутствует выпуклость в области IV и V пястно-фаланговых суставов; задержки умственного развития; нарушения минерального обмена (гиперфосфатемия, гипокальциемия, судороги, спазмы мышц, изменения скелета, в тяжелых случаях - патологические переломы, нарушение прорезывания зубов; эктопические кальцификаты - подкожные, в области базальных ганглиев). Выделяют два типа псевдогипопаратиреоза, отличающиеся биохимическими показателями: снижение содержания кальция в сыворотке крови, снижение экскреции фосфатов с мочой и повышение иммунореактивности паратиреоидного гормона плазмы наблюдается при обоих типах; экскреция цАМФ снижена при I и не изменена при II типе; концентрация фосфора в сыворотке повышена при I типе и не изменена или немного повышена при II типе. При цитогенетическом исследовании в ряде случаев выявляют микроделеции: сегмента q37 хромосомы 2 при I типе и сегмента q12 - q13.2 хромосомы 20. Тип наследования - предположительно Х-сцепленный доминантный для I типа; не установлен для II типа. Лечение: препараты кальция и витамина D в больших дозах; в случае выраженной деформации трубчатых костей - хирургическая коррекция.
F. Albright, C. H. Burnett, P. H. Smith, W. Parson. Pseudohypoparathyroidism: an example of «Seabright-Bantam syndrome». Endocrinology, Baltimore, 1942; 30: 922-932.
Найдено научных статей по теме — 4
Синдром Кушинга у новорожденного мальчика с синдромом маккьюна-олбрайта
Маказан Надежда Викторовна, Орлова Елизавета Михайловна, Карева Мария Андреевна, Поддубный Игорь Витальевич, Толстов Кирилл Николаевич, Полякова Галина Александровна, Богданова Полина Сергеевна, Петеркова Валентина Александровна, Дедов Иван Иванович
ВИПАДОК іЗ ПРАКТИКИ. ПОєДНАННЯ СИНДРОМУ МАК-КЮНА — ОЛБРАЙТА З АКРОМЕГАЛієЮ
СИНДРОМ МАК-КЬЮНА - ОЛБРАЙТА - БРАЙЦЕВА (ОБЗОР ЛИТЕРАТУРЫ И КЛИНИЧЕСКИЙ СЛУЧАЙ)
В статье приведены результаты обзора литературы, касающиеся синдрома Мак-Кьюна Олбрайта Брайцева, и описан клинический случай данного синдрома.
Роль молекулярно-генетических методов исследования в диагностике синдрома маккьюна-олбрайта-брайцева
Маказан Надежда Викторовна, Орлова Елизавета Михайловна, Колодкина Анна Александровна, Карева Мария Андреевна, Калинченко Наталья Юрьевна, Васильев Евгений Витальевич, Тюльпаков Анатолий Николаевич, Петеркова Валентина Александровна
Синдром МакКьюна-Олбрайта-Брайцева (МОБ) редкое генетическое заболевание, в основе которого лежат соматические мутации в гене GNAS.
Похожие термины:
Синдром Форбса-Олбрайта
по именам американских врачей A. P. Forbes и F. Albright, 1900-1969) - одно из эпонимических названий синдрома галактореи-аменореи, обусловленного аденомой гипофиза. A. P. Forbes, P. H. Henneman, G. C. Griswold, F. Albright. Syndrome characterize
БАТЛЕРА-ОЛБРАЙТА СИНДРОМ
описан американскими врачами A. Butler и F. Albright, 1900-1969; синоним - почечный тубулярный ацидоз, дистальный тип) - наследственное заболевание из группы тубулопатий, связанное с нарушением секреции ионо
Синдром Олбрайта-Хадорна
Первичное нарушение калиевого обмена с периодическими гипокалиемическими параличами мышц. Проявляется пароксизмами общей слабости, болями в костях, спине, остеопорозом костей, гипо- или арефле
МАРТИНА-ОЛБРАЙТА СИНДРОМ
Martin E., Albright F.]. Проявление нарушения утилизации тканями гормона паращитовидных желез. Характерны: гипокальциемия, гиперфосфатемия, ахондроплазия, умственное недоразвитие, судорожные тетанически
МАККЬЮНА-ОЛБРАЙТА СИНДРОМ
описан американскими педиатром D. J. McCune, 1902-1976, и эндокринологом F. Albright, 1900-1969; синоним - болезнь Олбрайта) - множественная фиброзная остеодисплазия с преждевременным половым развитием и пигмент
Синдром Олбрайта-Мак-Кьюна-Штернберга
Проявляется в детском возрасте. Этиология не известна. Проявляется нарушением функций шишковидной железы и других структур промежуточного мозга, что ведет к избытку продукции гонадотропинов и к
Псевдогипопаратиреоз. Остеодистрофия наследственная. Олбрайта синдром
Своеобразная остеодистрофия, проявляющаяся коротким туловищем, круглым лицом, укороченными метатарзальными, метакарпальными и фаланговыми костями, экзостозами. Олигофрения, эпилепсия, гиперкин
БАТЛЕРА-ОЛБРАЙТА СИНДРОМ
описан американскими врачами A. Butler и F. Albright, 1900-1969; синоним - почечный тубулярный ацидоз, дистальный тип) - наследственное заболевание из группы тубулопатий, связанное с нарушением секреции ионов водорода в просвет дистальных отделов извитых канальцев почек (либо с обратной диффузией их из просвета канальца в клетку). Развивается системный ацидоз с метаболическими нарушениями, приводящими к тяжелой гипокалиемии, гиперкальциурии. Проявления: отставание в росте, заторможенность, апатия, мышечная слабость, анорексия, рвота, жажда, полиурия, обезвоживание (следствие потери калия), поражение костей вследствие системного ацидоза (рахит у детей, остеомаляция с болями в костях и патологическими переломами у взрослых), нефрокальциноз, нефролитиаз с последующим присоединением инфекции мочевых путей. При обследовании выявляют метаболический ацидоз, гипокалиемию, гипонатриемию, щелочную реакцию мочи, гиперкальциурию. Тип наследования - аутосомно-доминантный или Х-сцепленный рецессивный. Лечение симптоматическое: коррекция ацидоза небольшими дозами бикарбонатов, препараты калия; при остеопорозе и остеомаляции - препараты витамина D и др.
A. Butler et al. Dehydration and acidosis with calcification of renal tubules. Journal of Pediatrics, St. Louis, 1936; 8: 489-499.
F. Albright, W. V. Consolazio, F. S. Coombs, J. H. Talbot, H. W. Sulkowitch. Metabolic studies and therapy in a case of nephrocalcinosis with rickets and dwarfism. Bulletin of the Johns Hopkins Hospital, Baltimore, 1940; 66: 7-33.
описан американскими врачами F. Albright, A. M. Butler и E. Bloomberg; синонимы - фосфат-диабет, семейный гипофосфатемический рахит, семейная Х-сцепленная гипофосфатемия, витамин D-резистентный рахит) - наследственное заболевание из группы врожденных тубулопатий, обусловленное ферментным дефектом, проявляющимся нарушением обратного всасывания фосфатов в проксимальных отделах почечных канальцев. Манифестирует обычно в конце 1-го - начале 2-го года жизни искривлением нижних конечностей у начинающих ходить детей. Клинические проявления напоминают рахит или остеомаляцию - задержка роста, деформация костей скелета, особенно голеней, ограничение подвижности в крупных суставах (тазобедренных, коленных, локтевых), из-за которой походка становится неуверенной, а в тяжелых случаях ребенок вообще перестает ходить. Отмечают компенсаторный поясничный лордоз, удлинение черепа в переднезаднем направлении (вследствие преждевременного окостенения стреловидного шва), болезненность костей и мышц. В отличие от витамин D-дефицитного рахита нет нарушения общего состояния, мышечной гипотонии, спазмофилии. Клинические проявления у гетерозиготных женщин выражены меньше, чем у больных мужчин. При лабораторном обследовании выявляют гипофосфатемию, повышение уровня щелочной фосфатазы при нормальных показателях кальция, гиперфосфатурию. При рентгенологическом исследовании отмечают рахитоподобые изменения костей скелета, грубоволокнистую структуру губчатого вещества костей. Тип наследования - Х-сцепленный доминантный. Применяют препараты витамина D в больших дозах (дозы, применяемые обычно для терапии рахита, неэффективны), фосфора, при грубых костных деформациях показана ортопедическая коррекция.
F. Albright, A. M. Butler, E. Bloomberg. Rickets resistant to vitamin D therapy. American Journal of Diseases of Children, Chicago, 1937; 54: 529-547.
Читайте также:
- Профилактика кариеса корня зуба. Чистка корней зуба при зубных камнях
- Случайные паразиты. Характеристика случайных паразитов в эпидемиологии.
- Функция сердца под действием полония. Влияние полония на костный мозг, селезенку
- Тамоксифен для лечения рака молочной железы у беременной
- Лучевая анатомия суставов, связок, мышц