Синдром Сфорцини (Sforzini) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 22.01.2026
Бесплатная консультация прямо сейчас!
Онлайн консультация специалиста по Вашему вопросу!
Номер лицензии: ЛО-77-01-019036
Что такое корсаковский синдром
Это нервно-психическое заболевание, основным симптомом которого является нарушение памяти. Оно обязано своим названием Сергею Корсакову, российскому психиатру, первым описавшим симптомы этого расстройства.
Заболевание вызвано дефицитом витамина B1, отвечающего за работу головного мозга. Слишком малое его количество в организме может повредить мозг, особенно те его части, которые отвечают за память. Они необратимы.
Из-за своей этиологии синдром Корсакова может возникать у людей, страдающих алкогольной зависимостью, поскольку длительное употребление алкоголя повреждает стенки кишечника, что снижает всасывание витамина B1 в желудочно-кишечном тракте. Расстройства пищевого поведения, включая анорексию и булимию, вызывают аналогичные симптомы. Заболевание также может возникать при раке кишечника и желудка, синдроме короткой кишки, опухолях головного мозга и механических повреждениях черепа.
Симптомы синдрома Корсакова
Заболевание проявляется рядом психических и неврологических симптомов.
В первую группу входят:
- Нарушения памяти: у пациента возникают трудности с запоминанием новой информации, при этом обычно нет трудностей с извлечением данных из долговременной памяти. Хотя могут быть проблемы с их упорядочиванием, расположением в хронологическом порядке. Пациент может путаться в фактах, неправильно интерпретировать события прошлого.
- Трудности с узнаванием людей. Человек в фазе психоза может испытывать трудности с распознаванием лиц и припоминанием их имен, даже тех, кого он хорошо знает (например, члены семьи).
- Проблемы с абстрактным мышлением, умозаключением, логическим мышлением.
- Нарушения сознания: у человека, страдающего синдромом Корсакова, могут быть трудности с определением времени, года, месяца, дня недели, времени, а также проблемы с припоминанием дат, например, своего рождения. Также могут наблюдаться трудности с восприятием пространства.
- Конфабуляции, то есть ложные воспоминания, призванные заполнить пробелы в памяти.
Вторая группа симптомов - неврологические - включают в себя:
- нарушения походки (дисбазия);
- нистагм, то есть частое непроизвольное движение глазными яблоками, а также другие проблемы со зрением;
- нарушения контроля движений и органов чувств;
- снижение или отсутствие реакции на свет;
- судорожные состояния.
В более тяжелых случаях могут быть повреждены периферические нервы и могут появиться кожные высыпания.
Онлайн консультация специалиста
по Вашему вопросу!
Диагностика и лечение синдрома Корсакова
Из-за обилия и разнообразия симптомов диагностировать заболевание непросто. Обычно диагностика включает в себя консультацию с пациентом, наблюдение за его поведением и медицинское обследование, в том числе проверку уровня витамина B1 в крови или моче.
Болезнь неизлечима. Для того, чтобы уменьшить выраженность симптомов и замедлить дегенеративные процессы, крайне важно как можно скорее начать лечение. Если человек находится в острой фазе алкогольного опьянения, должен быть произведен вывод из запоя под контролем специалистов.
Лечение заключается во введении витамина B1 в соответствующем количестве (50-100 мг в день, в зависимости от исходного уровня). Обычно его вводят внутривенно для максимально быстрого эффекта. Также необходимо выполнить компьютерную томографию, чтобы оценить, насколько большие изменения произошли в головном мозге. Однако главное в лечении - полностью отказаться от алкоголя.
Каков прогноз лечения? Все зависит от тяжести симптомов и степени повреждения мозга, вызванного дефицитом витамина B1. Подсчитано, что значительное облегчение симптомов возможно примерно у 25% пациентов. Примерно 50% людей могут рассчитывать на частичное выздоровление - обычно сохраняются некоторые проблемы с памятью. К сожалению, примерно у 25% пациентов симптомы не проходят.
Профилактика синдрома Корсакова
Прежде всего, избегайте употребления большого количества алкоголя. Этиловый спирт, точнее продукты его распада, повреждает пищеварительный тракт и препятствует усвоению витаминов и других полезных веществ. Также важно следить за уровнем витамина B1 в организме. Он содержится, среди прочего, в мясе, пшеничном хлебе, орехах и манной крупе.
Таким образом, синдром Корсакова - опасное неизлечимое заболевание, приводящее к необратимым изменениям в головном мозге, которые могут сохраняться всю жизнь. Его профилактика имеет важное значение.
Новое в лечении синдрома функционального перекреста
Функциональная патология, если исходить из практических ситуаций, то это комплекс функциональных расстройств, за которыми не кроется какая-то органическая патология. Причем, эта органическая патология должна быть обязательно исключена, и, только в таком случае мы можем говорить о функциональных изменениях, что заболевание обусловлено функциональными изменениями. Причем, в отличие от ситуации, которая уже не один десяток лет исповедовалась после открытия Вирхова, в данный момент никто не исключает именно то, что в основе этой функциональной патологии лежит какая-то дисфункция и структуры органа. Но теми исследованиями, которыми мы в настоящий момент обладаем, мы эти структурные изменения выявить не можем.
Надо сказать, что актуальность проблемы весьма высока. Дело в том, что в гастроэнтерологической практике от 40% до 60% лиц, обращающихся за помощью, имеют именно вот эту функциональную патологию. И, естественно, есть большой соблазн включить в группу функциональной патологии лиц, которые чем-то непонятны.
Но есть строгие правила. Обязательным исследованием является, если речь идет о жалобах со стороны кишечника, это, как минимум, ирригоскопия, а еще лучше - колоноскопия с биопсией. Обязательным исследованием является эзофагогастродуоденоскопия с осмотром Фатерова соска. И, конечно, ультразвуковое исследование брюшной полости.
При диагностике дисфункции желчевыводящих путей, под вопросом поставлено такое исследование, как ультразвуковая холецистография. Во врачебной практике к этому исследованию, наверное, следует скептически относиться в связи с тем, что оно занимает очень много времени, и не всегда его результаты соответствуют, в общем-то, тому, что имеет место на самом деле. И, кроме того, надо иметь в виду, что в динамике заболевания нередко одна форма дискинезии переходит в другую. Вот, как минимум, это необходимо провести.
Список заболеваний, которые включены в реестр функциональных расстройств, весьма высок. Это и заболевания пищевода, и заболевания желудка, главным образом функциональная диспепсия. Но особенно многогранные функциональные расстройства при патологии кишечника. И, конечно, главным среди этих расстройств, является синдром раздраженного кишечника.
На втором месте по обращаемости, видимо, стоят расстройства функции билиарного тракта. Они включают в себя расстройства желчного пузыря, и дисфункцию сфинктера Одди.
Что объединяет эти заболевания? Их, прежде всего, объединяет общий патогенез. Общий патогенез, который включает в себя нарушение висцеральной чувствительности, нарушение регулирования вегетативной нервной системы, нарушение чувствительности к гастроинтестинальным гормонам. И вот общий патогенез, объединяющий эти заболевания, позволил включить такое интересное явление, или такое интересное сочетание, как сочетание различных функциональных расстройств, которые объединены в понятие «синдром перекреста».
Дело в том, что это понятие пришло к нам из ревматологии, где также объединялись заболевания, имеющие общий патогенез. И примерно с 2005 года это понятие довольно прочно вошло в практику гастроэнтерологов. И, естественно, каждый раз подчеркивается, что главной причиной возникновение синдрома перекреста - это общие звенья патогенеза.
По обращаемости, после функциональной диспепсии, которая, вероятней всего, стоит на первом месте как функциональное расстройство желудочно-кишечного тракта, далее идет дискинезия желчевыводящих путей, или дисфункция желчевыводящих путей. Считается, что от 25% до 45% пациентов именно обладают этой патологией. Причем, обязательным условием, следует различать дисфункцию желчевыводящих путей вторичную, когда в основе лежит какое-то органическое страдание, это: желчекаменная болезнь, воспалительные заболевания. И первичную, когда в основе лежит именно нарушение функции. Поэтому, обязательным условием диагностики именно функциональных расстройств желчевыводящих путей, является тщательное обследование больного. На что можно ориентироваться при этом? Прежде всего, конечно, на болевой синдром. Болевой синдром, надо сказать, в первую очередь обусловлен у этих пациентов спазмом мускулатуры и самого желчного пузыря и желчных протоков. Что приводит к резкому повышению давления в билиарном тракте, так и к спастической ишемии. Спастическая ишемия неприятна не только тем, что она вызывает боль, но и тем, что вслед за ней может произойти дистрофия кишечной стенки, которая может усугубиться воспалением, образованием желчных полипов, и так далее. Также, в комплекс болевого синдрома входят и перерастяжение стенки желчного пузыря, вследствие реакции на спазм сфинктера Одди, что, естественно, приводит к ретроградному повышению давления. Помимо этого, в комплекс патологии желчевыводящих путей функционального генеза, входит желудочная диспепсия, обусловленная нарушением поступления желчи 12-перстную кишку, что приводит к ацидитации 12-перстной кишки. И большую роль, конечно, играет вегетативная дисфункция, эмоциональная лабильность и системные реакции, которые обусловлены недостатком желчи, которые приводят к синдрому избыточного бактериального роста в кишечнике, и так далее. Но надо сказать, что «синдром перекреста» ни в коем случае не подразумевает включение патологии, которая является осложнением того, или иного заболевания. Это, прежде всего, два независимых заболевания.
И важнейшим из функциональных расстройств является, конечно, дисфункция сфинктера Одди. Судя по тем синонимам, которые здесь представлены, это: и дискинезия желчевыводящих путей, и гипертоническая дискинезия сфинктера Одди, спазм сфинктера Одди, прежде всего клиницисты, исследователи обращают внимание именно на спазм тех структур, которые входят в понятие сфинктера Одди. И, в связи с этим, обращено внимание на то, какую роль в развитии дисфункции сфинктера Одди играет важнейший гормон, который определяет моторику желчевыводящей системы, это, конечно, холецистокинин. Холецистокинин обязательно участвует в процессе сокращения мышц желчного пузыря. Но, одновременно, для того, чтобы все шло адекватно, и желчь адекватно выделялась, при повышении давления в желчном пузыре, в 12-перстную кишку, он же, холецистокинин, способствует расслаблению сфинктера Одди. Эпидемия, фактически, эпидемия желчекаменной болезни привела к тому, что холецистэктомия стала, после аппендэктомии, самой распространенной операцией. В США до 500 тыс. пациентов получают такое вспоможение, в России несколько меньше. И, как результат этого вмешательства, происходит следующее. Произошла холецистэктомия, нет желчного пузыря. А в желчном пузыре выделяется гормон, который является антагонистом холецистокинина. Он как раз снижает воздействие холецистокинина на желчный пузырь, и на сфинктер Одди. В связи с тем, что нет этого гормона гастроинтестинального, происходит повышение сфинктера Одди. Поэтому так называемый постхолецистный синдром - это, прежде всего, повышение тонуса сфинктера Одди.
Клиническими проявлениями дисфункции сфинктера Одди являются рецидивирующие приступы болей в эпигастральной области и в правом подреберье. Эти боли длятся более 20 минут, и, самое главное, что они повторяются. Ну, и в анамнезе, конечно, либо холецистэктомия, либо в редких случаях дисфункция сфинктера Одди бывает и при сохраненном желчном пузыре, но это не так часто встречается. Дисфункция сфинктера Одди, помимо клинических проявлений, которые заставляют обратиться к врачу, обязательно должна быть диагностирована по варианту. Прежде всего, это билиарный вариант. Этот вариант обусловлен тем, что повышается преимущественно давление в билиарном тракте. Он сопровождается болями в правом подреберье, и, что очень важно, повышением АЛТ, АСТ, гамма-глютаминтранспептидазы, щелочной фосфатазы. Но очень важным является то обстоятельство, что после стихания болей этот биохимический синдром уходит. При панкреатическом варианте с диагностикой несколько сложнее, поскольку здесь крайне трудно отличить панкреатический вариант дисфункции сфинктера Одди от атаки серозного такого, нетяжелого панкреатита, поскольку в критерии входят повышение амилазы и боли, похожие на панкреатические. Естественно, что диагностика панкреатического варианта более ответственна, и она уже требует и проведения эндоскопической ретроградной холангиопанкреатографии, когда будет доказано замедление выделения контрастного вещества, и компьютерной томографии для того, чтобы четко определить состояние поджелудочной железы.
Третьим по встречаемости является, конечно, синдром раздраженного кишечника. Синдром раздраженного кишечника - это обязательно боль, которая сочетается с нарушением характера кала. И в основе практически всего комплекса болей при синдроме раздраженного кишечника входит именно спастическое сокращение мышц кишки. И это объединяет дисфункцию сфинктера Одди, дискинезию желчного пузыря с этим синдромом, поскольку в основе и того, и другого лежит именно спазм гладкой мускулатуры желудочно-кишечного тракта.
Помимо перечисленных на слайде симптомов, к симптомам, подтверждающим диагноз «синдром раздраженного кишечника» входят и симптомы, обусловленные ускорением моторики, такие, как: императивные позывы, и эффект внезапного опорожнения кишечника, так называемая, «медвежья болезнь», и выделение слизи, что подчеркивает, что это не только нарушение моторики, но и нарушение секреции, и чувство переполнения, переливания, что обусловлено, конечно, нарушением висцеральной чувствительности. Так же, как и при дискинезии сфинктера Одди, в основе болей при синдроме раздраженного кишечника лежит спазм и растяжение. Надо сказать, что общий патогенез, в основе которого лежит, конечно, как эффект всех привходящих факторов, спазм гладкой мускулатуры, приводит к тому, что эти заболевания встречаются совместно. Вот, в самом раннем таком исследоваии было выявлено, что у афроамериканцев, при синдроме абдоминальной боли, сочетание синдрома раздраженного кишечника и дисфункции сфинктера Одди было выявлено в 7%. Однако, при нацеленном исследовании сибирские авторы Осипенко и его коллеги показали, что у больных синдромом раздраженного кишечника при ультразвуковом и манометрическом исследованиях спазм сфинктера Одди был выявлен более чем в 40%. И такое более нацеленное изучение этого эффекта показало, что сочетание синдрома раздраженного кишечника и дискинезии желчевыводящих путей, то есть, перекрест этих двух заболеваний, сопровождается более выраженными абдоминальными болями, то есть, сложением болей в правом подреберье и в области толстой кишки, заметным повышением активности биохимических маркеров холестаза, что, наверняка, связано со спазмом и повышением давления в 12-перстной кишке. А также повышением тревожности, повышением уровня депрессии у этих больных, что ведет, конечно, к значительному снижению показателей жизни. В основе и того, и другого лежит, конечно, прежде всего, висцеральная гиперчувствительность, которая обусловлена либо снижением порога болевой чувствительности, видимо, обусловленная нарушением метаболизма серотонина и эндорфинов, и, конечно, гиперчувствительностью к воздействию гастроинтестинальных гормонов. Надо сказать, что очень схожие результаты психиатрического исследования получены как при исследовании пациентов с синдромом раздраженного кишечника, и, как вы видите, здесь довольно высокий процент пациентов с аффективными расстройствами, так и, могу сказать, что точно такая же примерно картина была получена и при исследовании больных с дискинезией желчевыводящих путей.
В основе терапии пациентов, у которых сочетаются эти, как минимум два функциональных заболевания, лежит, прежде всего, коррекция диеты. Она целиком зависит от варианта нарушения функции кишечника, и, естественно, диета должна учитывать обязательно состояние желчевыводящих путей. Поскольку чаще всего болевым синдромом сопровождаются именно гиперкинетическая дискинезия желчного пузыря или спазм сфинктера Одди, то, несмотря на то, что при СРК с запорами рекомендуют увеличить количество растительных жиров, просто жиров, поскольку это улучшает перистальтику кишечника, здесь, конечно, к этому вопросу надо отнестись с большой настороженностью. Диета должна быть механически щадящая. И совершенно позитивно на функцию желчного пузыря, сфинктера Одди и кишечника влияет назначение пшеничных отрубей и других растительных волокон. И, наконец, вторым важнейшим моментом является борьба с главным виновником всей клинической картины - это купирование болевого синдрома. Прежде всего, спазмолитики. Естественно, нельзя обойтись без нормализации функции центральной нервной системы и вегетативной нервной системы - это психотропные средства, которые включают антидепрессанты, трициклические, или же ингибиторы обратного захвата серотонина, и иногда даже речь идет о нейролептиках.
И, естественно, этим больным, особенно с синдромом раздраженного спазма сфинктера Одди, нецелесообразно назначать ферментные препараты, содержащие желчь, а следует обойтись пищеварительными ферментами, не содержащими желчь. В основе борьбы с болевым синдромом лежит назначение спазмолитиков. Это препараты первого выбора при болях у пациентов с синдромом функционального перекреста. И надо сказать, что в этом случае спазмолитик - это не симптоматическая терапия, а это компонент патогенетической и даже этиопатогенетической терапии, поскольку спазм несет в себе фактически начало всех начал: и боль, и нарушение функции, и так далее. Поэтому, надо решить, какие же спазмолитики целесообразно назначить. Конечно, нецелесообразно при синдроме перекреста назначать М-холинолитики, учитывая, что у этих больных очень часто клиническая картина гастроэнтерологическая сочетается и с нейроциркуляторной дистонией, и с тахикардией, и так, далее, поэтому, целесообразно назначить миотропные спазмолитики. И здесь, в общем-то, выбор за врачом.
Принципиальные спазмолитики - это и ингибиторы фосфодиэстеразы, блокаторы кальциевых каналов, блокаторы натриевых каналов. И имеет смысл обратить внимание на препараты, которые оказывают свой эффект, воздействуя на фосфодиэстеразу. Это связано с тем, что этот путь воздействия на спазм гладкой мышцы фактически приводит к одинаковому эффекту в значительных участках желудочно-кишечного тракта. Одновременное воздействие этой группы спазмолитиков, оказывается и в области кишечника, и билиарного тракта, естественно, и в области мочевыводящих путей. При этом конечно есть, или, по-крайней мере, обсуждалась опасность, что эта группа препаратов может оказать влияние на тонус сосудов. В связи с этим следует обратить внимание, что не все изомеры фосфодиэстеразы действуют на гладкую мускулатуру. Вот, фосфодиэтераза V действует на сосуды, фосфодиэстераза III - на кардиомиоциты, а используемая нами в клинической практике фосфодиэстераза IV как раз и действует только на гладкую мускулатуру как сфинктера Одди, так и на гладкую мускулатуру кишечника. При этом следует обратить внимание, что одновременно этот эффект сопровождается и борьбой с отеком, то есть, воздействует на отек, и, также, воздействует на воспаление. В связи с этим также есть благотворное воздействие на болевой синдром, метеоризм и нарушение стула.
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма
Профессиональные диагностические инструменты. Оценка эластичности тканей, расширенные возможности 3D/4D/5D сканирования, классификатор BI-RADS, опции для экспертных кардиологических исследований.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) - это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза-Франческетти, синдром Франческетти-Цвалена-Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер - имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза-Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
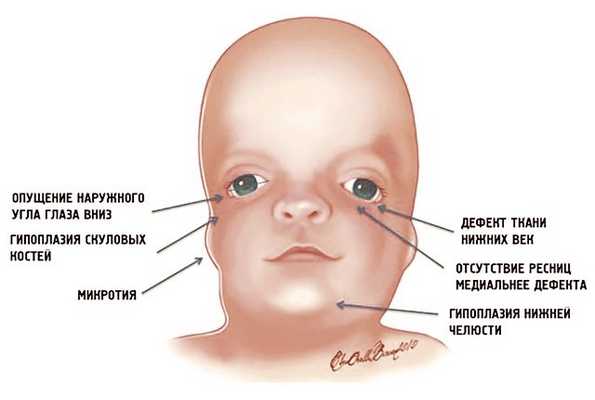
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса 2, также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости 4. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев - это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 - q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4-6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип - мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши - все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности - редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким - 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12-13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2-4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
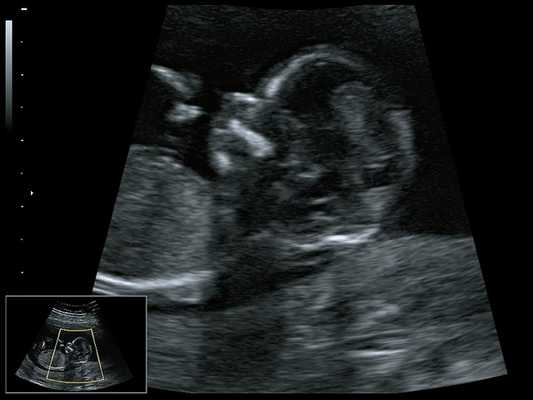
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
Синдром Тричер Коллинза
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
При внутриутробных нарушениях процессов развития костей возникают серьезные черепно-лицевые деформации, и одной из разновидностей такой патологии является синдром Тричер Коллинза (TCS) или мандибулофасциальный, то есть челюстно-лицевой дизостоз.
Код заболевания по МКБ 10: класс XVII (врожденные аномалии, деформации и хромосомные нарушения), Q75.4 - mandibulofacial dysostosis.
Код по МКБ-10
Эпидемиология
Распространенность синдрома Тричер Коллинза находится в диапазоне - один случай на 25- 50 тыс. новорожденных обоего пола (хотя британские медики отмечают, что патология выявляется у одного младенца из 10-15 тыс.).
Причины синдрома Тричер Коллинза
Данный синдром получил имя выдающегося британского офтальмолога Эдварда Тричер Коллинза, описавшего основные черты патологии более ста лет назад. Однако европейские врачи чаще называют этот вид аномалии костей лица и челюстей болезнью или синдромом Франческетти - на основании обширных исследований швейцарского офтальмолога Адольфа Франческетти, который ввел термин «мандибулофасциальный дизостоз» в середине прошлого века. В медицинских кругах также используется название - синдром Франческетти-Коллинза.
Причины синдрома Тричер Коллинза - мутации гена TCOF1 (в локусе хромосомы 5q31.3-33.3), который кодирует ядрышковый фосфопротеин, отвечающий за формирование черепно-лицевой части эмбриона человека. В результате преждевременного уменьшения количества этого белка нарушаются биогенез и функции рРНК. По мнению генетиков исследовательской программы Human Genome, эти процессы приводят к сокращению пролиферации эмбриональных клеток нервного гребня - валика вдоль нервного желоба, который в ходе развития зародыша замыкается в нервную трубку.
Формирование тканей лицевой части черепа происходит благодаря трансформации и дифференциации клеток верхней (головной) части нервного гребня, которые мигрируют вдоль нервной трубки в область первой и второй жаберных дуг зародыша. И дефицит этих клеток вызывает черепно-лицевые деформации. Критический период возникновения аномалий - с 18 по 28 день после оплодотворения. По завершении миграции клеток нервного гребня (на четвертой неделе гестации) образуются практически все рыхлые мезенхимальные ткани в области лица, которые позже (с 5 по 8 недели) дифференцируются в скелетные и соединительные ткани всех частей лица, шеи, гортани, уха (в том числе внутреннего) и будущих зубов.
Патогенез
Патогенез синдрома Тричер Коллинза часто имеет семейный характер, и аномалия наследуется по аутосомно-доминантному принципу, хотя бывают случаи аутосомно-рецессивной передачи дефекта (при мутациях других генов, в частности, POLR1C и POLR1D). Самым непредсказуемым в челюстно-лицевом дизостозе является то, что мутация наследуется детьми только в 40-48% случаев. То есть у 52-60% пациентов причины синдрома Тричер Коллинза не связаны с наличием аномалии в роду, и, как полагают, патология возникает в результате спорадических генных мутаций de novo. Вероятнее всего, новые мутации представляют собой последствия тератогенного воздействия на плод во время беременности.
В числе тератогенных причин данного синдрома специалисты называют большие дозы этанола (этилового спирта), радиацию, сигаретный дым, цитомегавирус и токсоплазму, а также гербициды на основе глифосата (Раундал, Глифор, Торнадо и др.). А в список ятрогенных факторов попали препараты от угрей и себореи с 13-цис-ретиноевой кислотой (Изотретиноин, Аккутан); противосудорожное лекарство Фенитоин (Дилантин, Эпанутин); психотропные средства Диазепам, Валиум, Реланиум, Седуксен.
Симптомы синдрома Тричер Коллинза
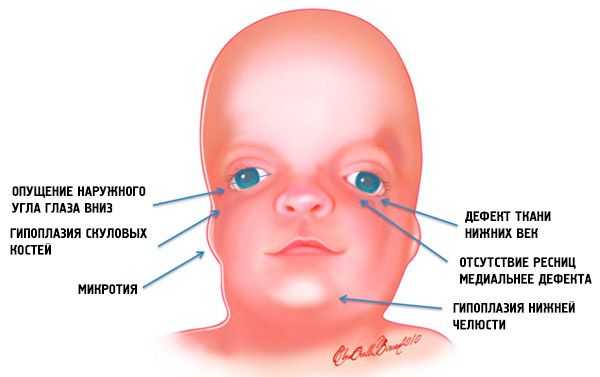
По большей части, клинические признаки мандибулофасциального дизостоза и степень их выраженности зависят от особенностей проявления генных мутаций. И первые признаки данной аномалии в большинстве случаев видны у ребенка сразу же после его появления на свет: лицо при синдроме Тричер Коллинза имеет характерный вид. Причем морфологические аномалии обычно двусторонние и симметричные.
Наиболее очевидные симптомы синдрома Тричер Коллинза:
- недоразвитость (гипоплазия) лицевых костей черепа: скуловых, скуловых отростков лобной кости, боковых крыловидных пластинок, придаточных пазух носа, нижней челюсти и выступов костных эпифизов (мыщелков);
- недоразвитие костей нижней челюсти (микрогнатия) и более тупой чем обычно нижнечелюстной угол;
- нос имеет нормальный размер, однако, кажется большим из-за гипоплазии надбровных дуг и недоразвитости или отсутствия скуловых дуг в области висков;
- глазные щели нисходящие, то есть разрез глаз аномальный, с опущенными вниз наружными уголками;
- дефекты нижних век (колобома) и частичное отсутствие ресниц на них;
- ушные раковины неправильной формы с широким диапазоном отклонений, вплоть до их расположения в углу нижней челюсти, отсутствия мочек, слепых свищей между козелком уха и углом рта и др.;
- сужение или заращение (атрезия) наружного слухового каналов и аномалии косточек среднего уха;
- отсутствие или гипоплазия околоушных слюнных желез;
- фарингеальная гипоплазия (сужение глотки и дыхательных путей);
- несращение твердого нёба (волчья пасть), а также отсутствие, укорочение или неподвижность мягкого неба.
Такие анатомические аномалии во всех случая имеют осложнения. Это функциональные нарушения слуха в виде проводящей (кондуктивной) тугоухости или полной глухоты; нарушения зрения из-за неправильно формирования глазных яблок; дефекты нёба вызывают трудности с кормлением и глотанием. Имеются связанные с дефектами челюстей нарушения окклюзии зубов (неправильный прикус), что, в свою очередь, вызывает проблемы с жеванием и артикуляцией. Патологии мягкого неба объясняют гнусавость голоса.
Осложнения и последствия
Последствия челюстно-лицевых аномалий при синдроме Тричер Коллинза проявляются в том, что при рождении ребенка его интеллектуальные способности нормальные, но из-за дефектов слуха и других нарушений отмечается вторичная задержка умственного развития.
Кроме того, дети с такими дефектами остро чувствуют свою ущербность и страдают, что негативно сказывается на их нервной системе и психике.
Диагностика синдрома Тричер Коллинза
Постнатальная диагностика синдрома Тричер Коллинза, по существу, проводится на основании клинических признаков. Челюстно-лицевой дизостоз легко определяется при полный экспрессивности синдрома, но когда присутствуют минимально выраженные симптомы патологии, с постановкой правильного диагноза могут возникнуть проблемы.
При этом особого внимания требует оценка всех связанных с аномалиями функций, особенно тех, что затрагивают дыхание (в связи с угрозой апноэ во сне). Также проводится оценка и мониторинг эффективности кормления и насыщения гемоглобина кислородом.
В дальнейшем - на 5-6 день после рождения - предстоит выяснить степень повреждений слуха с помощью аудиологического тестирования, которое должно проводиться еще в родильном доме.
Назначается обследование, в ходе которого инструментальная диагностика проводится рентгеноскопией черепно-лицевой дисморфологии; пантомографией (панорамным рентгеном костных структур лицевого черепа); полной черепной компьютерной томографией в различных проекциях; КТ или МРТ головного мозга для определения состояния внутреннего слухового прохода.
Самое раннее - пренатальное - диагностирование челюстно-лицевых аномалий при наличии синдрома Тричер Коллинза в семейном анамнезе возможно путем биопсии ворсин хориона на 10-11 неделе беременности (процедура угрожает выкидышем и занесением инфекции в матку).
Но чаще всего в дородовой диагностике данного синдрома у плода используется УЗИ (на 20-24 неделях беременности).
Синдром Дресслера
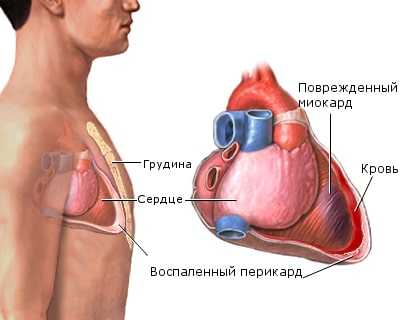
Постинфарктный синдром (или синдром Дресслера) — реактивное аутоиммунное осложнение инфаркта миокарда, развивающееся через 2—6 недель после его начала.
Частота развития
Первоначально считалось, что синдром Дресслера возникает примерно у 4% больных, перенесших инфаркт миокарда. C учетом атипичных и малосимптомных форм частота его развития значительно выше - 15-23 %, а по некоторым источникам достигает 30 %. Однако в последние годы частота синдрома Дресслера уменьшилась. Причинами могут быть широкое использование нестероидных противовоспалительных средств (ацетилсалициловой кислоты) и распространение реперфузионных методов лечения ИМ, уменьшающих объем повреждения мышцы сердца. Другой причиной снижения частоты развития синдрома Дресслера может быть включение в комплекс терапии инфаркт миокарда ингибиторов ангиотензинпревращающего фермента, антагонистов альдостерона и статинов, вследствие их иммуномодулирующего и противовоспалительного действия. Постинфарктый синдром развивается в подостром периоде (не ранее 10-го дня от момента заболевания) у 3—4 % пациентов, перенёсших инфаркт миокарда.
Причины развития
Основная причина синдрома Дресслера - инфаркт миокарда. Считается, что синдром Дресслера чаще развивается после крупноочаговых и осложненных инфарктов, а также после кровотечений в полость перикарда. Синдром Дресслера, точнее синдром постповреждения сердца, может развиваться после кардиохирургических вмешательств (постперикардиотомический синдром, посткомиссуротомический синдром). Помимо этого, типичные признаки синдрома Дресслера могут появляться после других повреждений сердца (ранение, контузия, непроникающий удар в область грудной клетки, катетерная абляция). В настоящее время синдром Дресслера рассматривается как аутоиммунный процесс, обусловленный аутосенсибилизацией к миокардиальным и перикардиальным антигенам. Определенное значение придается также антигенным свойствам крови, попавшей в полость перикарда.
При постинфарктном синдроме наблюдаются изменения и в клеточном иммунитете. Так, имеются данные, что при синдроме Дресслера значительно повышен уровень цитотоксических T-клеток. Этиологическим фактором синдрома Дресслера может быть инфекция, в частности вирусная, поскольку у больных, у которых этот синдром развился после кардиохирургических вмешательств, часто регистрируют повышение титра противовирусных антител.
Симптомы и течение
Развивается на 2-4-й неделе инфаркта миокарда, однако эти сроки могут уменьшаться - «ранний синдром Дресслера» и увеличиваться до нескольких месяцев, «поздний синдром Дресслера». Иногда течение синдрома Дресслера принимает агрессивный и затяжной характер, он может длиться месяцы и годы, протекать с ремиссиями и обострениями. Основные клинические проявления синдрома: лихорадка, перикардит, плеврит, пневмонит и поражение суставов. Лихорадка при синдроме Дресслера не имеет какой-либо строгой закономерности. Как правило, она бывает субфебрильной, хотя в отдельных случаях может быть фебрильной или вообще отсутствовать.
Перикардит является обязательным элементом синдрома Дресслера. Клинически он проявляется болью в перикардиальной зоне, которая может иррадиировать в шею, плечо, спину, брюшную полость. Боль может быть острой приступообразной (плевритическая) или давящей, сжимающей (ишемической). Она может усиливаться при дыхании, кашле, глотании и ослабевать в вертикальном положении или лежа на животе. Она длительная и исчезает или ослабевает после появления в полости перикарда воспалительного экссудата. Главный аускультативный признак перикардита - шум трения перикарда: в первый день болезни при внимательной аускультации он определяется у абсолютного большинства (до 85 %) больных. Шум лучше всего выслушивается у левого края грудины, при задержке дыхания и наклоне туловища пациента вперед. В классическом варианте он состоит из трех компонентов - предсердного (определяется в систолу) и желудочкового (систолического и диастолического). Как и боль, шум трения перикарда уменьшается или исчезает вовсе после появления в полости перикарда выпота, раздвигающего трущиеся листки перикарда. Обычно перикардит протекает нетяжело: уже через несколько дней боли стихают, а экссудат в полости перикарда почти никогда не накапливается в таком количестве, чтобы ухудшить кровообращение, хотя иногда могут появиться признаки тяжелой тампонады сердца. Иногда воспалительный процесс в перикарде при синдроме Дресслера принимает затяжной рецидивирующий характер и заканчивается развитием констриктивного перикардита. При применении антикоагулянтов на фоне синдрома Дресслера возможно также развитие геморрагического перикардита, хотя подобное осложнение может быть и при отсутствии антикоагулянтной терапии.
Плеврит. Проявляется болью в боковых отделах грудной клетки, усиливающейся при дыхании, затруднением дыхания, шумом трения плевры, притуплением перкуторного звука. Он может быть сухим и экссудативным, односторонним и двусторонним. Нередко плеврит носит междолевой характер и не сопровождается типичными физикальными симптомами.
Пневмонит. Пневмонит при синдроме Дресслера выявляется реже, чем перикардит и плеврит. Если очаг воспаления достаточно велик, также отмечается притупление перкуторного звука, ослабленное или жесткое дыхание, появление фокуса мелкопузырчатых хрипов. Возможен кашель и выделение мокроты, иногда с примесью крови, что всегда вызывает определенные диагностические трудности.
Поражение суставов. Для синдрома Дресслера характерно появление так называемого «синдрома плеча»: болезненных ощущений в области плечелопаточных суставов, чаще слева, ограничение подвижности этих суставов. Вовлечение в процесс синовиальных оболочек нередко приводит к возникновению болей и в крупных суставах конечностей.
Другие проявления. Проявлением постинфарктного синдрома может быть сердечная недостаточность вследствие диастолической дисфункции, геморрагический васкулит и острый гломерулонефрит.
Методы исследования
Лабораторные данные. Часто отмечается повышение СОЭ и лейкоцитоз, а также эозинофилия. Весьма характерно резкое повышение уровня С-реактивного белка. У больных с синдромом Дресслера регистрируются нормальные уровни маркеров повреждения миокарда (МВ-фракция креатинфосфокиназы (МВ-КФК), миоглобин, тропонины), хотя иногда отмечается их незначительное повышение, что требует проведения дифференциальной диагностики с рецидивом инфаркта миокарда.
Электрокардиография (ЭКГ). При наличии перикардита на ЭКГ определяются диффузный подъем сегмента ST и, периодически, депрессия сегмента PR, за исключением отведения aVR, в котором наблюдаются депрессия ST и подъем PR. По мере накопления экссудата в полости перикарда может снизиться амплитуда комплекса QRS.
Эхокардиография. При накоплении жидкости в полости перикарда выявляется сепарация его листков и могут появиться признаки тампонады сердца. Для синдрома Дресслера не характерен большой объем жидкости в полости перикарда - как правило, сепарация листков перикарда не достигает 10 мм в диастолу.
Рентгенография. Обнаруживают скопление жидкости в плевральной полости, междолевой плеврит, расширение границ сердечной тени, очаговые тени в легких.
Компьютерная или магнитнорезонансная томография также выявляют жидкость в полости плевры или перикарда и легочную инфильтрацию.
Плевральная и перикардиальная пункция. Извлеченный из полости плевры или перикарда экссудат может быть серозным или серозно-геморрагическим. При лабораторном исследовании в нем определяется эозинофилия, лейкоцитоз и высокий уровень С-реактивного белка.
Лечение
Нестероидные противовоспалительные препараты (НПВС). Препаратом выбора при синдроме Дресслера традиционно считается ибупрофен (400- 800 мг/сут). Реже используют аспирин. Хирургическое лечение применяется при констриктивном перикардите.
Осложнения
Тампонада сердца, геморрагический или констриктивный перикардит, окклюзия (сдавление) коронарного шунта и редко - анемия.
Прогноз
Прогноз при синдроме Дресслера, как правило, благоприятный. Вместе с тем его течение иногда принимает затяжной рецидивирующий характер. Кроме того, имеются данные о том, что выживаемость в течение 5 лет среди перенесших этот синдром, хотя и незначительно, но снижается.
Аутоиммунный процесс развивающийся после инфаркта миокарда и проявляющийся перикардитом, плевритом, воспалительными заболеваниями суставов и сосудов. Лечение проводится с помощью противовоспалительных препаратов.
Читайте также:
- Влияние инсулина на обмен углеводов. Обмен глюкозы под действием инсулина
- Эккринная порома
- Пример двустороннего пневмоторакса. Клиника клапанного двустороннего пневмоторакса
- Раннее выявление злоупотребления алкоголем. Скрининг населения на злоупотребление алкоголем.
- Антибактериальные средства. Антибактериальные препараты. Антибиотики. Классификация антибиотиков.