УЗИ, МРТ при синдроме Экарди у плода
Добавил пользователь Alex Обновлено: 08.01.2026
Что представляют собой ВПР (врожденные пороки развития)? Врожденные пороки развития - это одна из основных причин перинатальной смертности и инвалидности детского населения. Среди аномалий развития особую значимость имеют пороки развития, затрагивающие центральную нервную систему плода.
Поэтому важнейшей задачей настоящего времени является максимально раннее выявление врожденных пороков развития. Своевременное диагностирование патологий развития плода позволяет контролировать процесс уже на ранних этапах беременности, выбирать необходимую тактику ее ведения, уточнять сроки и способ родоразрешения, при необходимости проводить хирургическую или терапевтическую коррекцию, а в тяжелых случаях решать вопрос о прерывании беременности.
Что выбрать: УЗИ или МРТ плода? Основным методом диагностики врожденных пороков развития в современном акушерстве является УЗИ - диагностика.
Зачем и как проводится гинекологическое УЗИ? Рассказывает врач ультразвуковой диагностики «Клиника Эксперт Оренбург» Поскребышева Анна Викторовна
Проведение скрининговых исследований играет важнейшую роль в выявлении аномалий развития. Однако в тех случаях, когда возможности сонографии ограничены (ожирение матери, олигогидрамнион, ягодичное предлежание плода, редкая патология плода и др.), дополнительным методом исследования состояния плода является МРТ-исследование.
Зачем делают МРТ беременным? Читать далее
Метод МРТ: обладает высокой разрешающей способностью в визуализации структур головного мозга, позволяет получить мультипланарные изображения с большим полем обзора, не связан с ионизирующим излучением - безвреден; МРТ плода позволяет не только уточнять характер аномалий, заподозренных на УЗИ, но и выявлять ту патологию, определение которой находится за пределами возможностей сонографии.
В каких случаях МРТ противопоказано? Рассказывает исполнительный директор «Клиника Эксперт Оренбург» Подлевских Юрий Андреевич
Рассмотрим несколько примеров МРТ-исследования плода.
На рисунке - тазовое предлежание плода, 26 недель. Нормальная анатомия головного мозга
На рисунке - плод, 38 недель. Структурные изменения головного мозга плода не выявлены. Определяется однократное обвитие пуповиной
На рисунке - плод, 39 недель. Двукратное обвитие пуповиной
На рисунке - плод, 34 недели. Головное предлежание, гипоплазия мозжечка
Диагностика мальформаций мозолистого тела по данным УЗИ часто представляет сложности. При этом результаты МРТ-исследования уже на 20 неделе гестации сравнимы с данными МРТ в постнатальный период.
На рисунке - плод, 29 недель. Агенезия мозолистого тела. Асимметричная гидроцефалия
МРТ плода позволяет диагностировать такие нарушения нейрональной миграции, как лисэнцефалия, гетеротопия, шизэнцефалия, мало доступные для диагностики при других методах исследования.
На рисунке - плод, 32 недели; поперечное положение плода. Шизэнцефалия
Методом УЗИ-скрининга также возможно выявление черепно-мозговых грыж плода. Но при этом точно определить содержимое грыжевого мешка возможно только при МРТ-исследовании.
На рисунке - плод, 23 недели. Теменно-затылочное энцефалоцеле, заполненное спинномозговой жидкостью без признаков вовлечения мозговой ткани
Метод МРТ имеет большое значение в выявлении не только пороков развития ЦНС, но и при заболеваниях и аномалиях паренхиматозных органов брюшной полости и малого таза плода, его опорно-двигательного аппарата, крупных сосудов и др.
На рисунке - плод, 22 недели. Энтерогенная киста
На рисунке - врожденная диафрагмальная грыжа
На рисунке - мультикистоз почки
На рисунке - плод, 28 недель. Атрезия пищевода
На рисунке - пупочная грыжа
Таким образом, МРТ является дополнительным способом визуализации структур головного мозга плода. Он дополняет ультразвуковое исследование в тех случаях, когда применение сонографии технически ограничено, либо ее результаты сомнительны и противоречивы.
УЗИ, МРТ при синдроме Экарди у плода
Российская медицинская академия последипломного образования;
Тушинская детская городская больница, Москва
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7): 95‑97
Милованова О.А. Синдром Айкарди. Журнал неврологии и психиатрии им. С.С. Корсакова. 2011;111(7):95‑97.
Milovanova OA. Aicardi syndrome. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2011;111(7):95‑97. (In Russ.).
Синдром Айкарди - врожденное мультисистемное заболевание неясной этиологии, характеризующееся тетрадой симптомов: эпилептическими приступами по типу инфантильных спазмов, частичным или полным отсутствием мозолистого тела, хориоретинальными лакунарными очагами, умственной отсталостью [1, 15, 21, 31, 32, 34]. Впервые заболевание было описано J. Aicardi в 1958 г. [4]. Всего в мире отечественными и зарубежными авторами описано 200 случаев синдрома Айкарди [1, 4, 11]. Считается, что синдром Айкарди - Х-сцепленное доминантное заболевание, приводящее к летальному исходу у лиц мужского пола [22, 26]. J. Holmes и соавт. [16] описали единственного мальчика с XXY-кариотипом. Некоторые исследователи предполагали, что все известные случаи заболевания являются результатами хромосомных мутаций, однако сведения о пораженных сибсах отсутствовали [11]. Не исключено, что эмбриональный мозаицизм при мутации генов может быть наследственно обусловленным [20]. H. Ropers и соавт. [26] выявили микроделецию в Хр22.3 у девочки с синдромом Айкарди. Предполагается, что патогенез данного заболевания связан с генетически детерминированными нарушениями на этапе нейрональной миграции. Морфологическое исследование головного мозга подтверждает множественные аномалии его развития, включающие полную или частичную агенезию мозолистого тела, гетеротопию коркового вещества мозга, аномалии строения извилин (чаще по типу микрогирии и гетеротопии), нарушения клеточной архитектоники.
Наиболее яркими клиническими проявлениями синдрома Айкарди являются неврологические расстройства. Эпилептические приступы в виде инфантильных спазмов доминируют у детей 1-го года жизни. По классификации О. Dulac и соавт. [12] различают флексорные, экстензорные, флексорно-экстензорные, тонические или миоклонические, симметричные или асимметричные, синхронные или асинхронные, с фокальным компонентом, серийные или единичные инфантильные спазмы. У детей с синдромом Айкарди чаще выявляются латерализованные флексорные инфантильные спазмы, сочетающиеся у части больных с другими видами эпилептических приступов (чаще фокальными). Установлено, что чем меньше возраст дебюта приступов при данном заболевании, тем тяжелее степень нарушения психомоторного развития [6, 8, 10].
У детей с синдромом Айкарди часто встречаются черепно-лицевые дисморфии (микро-, гидроцефалия, аномалии ушной раковины), реже - вертебральные и реберные мальформации.
Практически у всех детей с синдромом Айкарди выявляются нарушения психомоторного развития различной степени. В случаях с задержкой психомоторного развития, предшествующей дебюту эпилептических приступов, неврологический регресс в дальнейшем усугубляется [23]. Отсутствует интерес к окружающему миру, потерян контакт с ребенком, снижается дифференцировка эмоциональных реакций, практически прекращается предречевое развитие. Постепенно развивается двигательный дефект, в ряде случаев до степени тетрапареза центрального генеза с полным отсутствием развития двигательных навыков и установочных рефлексов. В 90% наблюдений нарушаются познавательные функции [25, 35].
В ряде случаев метод ультразвукового сканирования плода позволяет выявить наличие синдрома Айкарди на ранних стадиях гестационного развития.
При визуализационных методах - нейросонографии (НСГ), рентгеновской компьютерной томографии (КТ), магнитно-резонансной томографии (МРТ) головного мозга - патогномоничным признаком синдрома Айкарди является агенезия мозолистого тела. Выделяют полную агенезию мозолистого тела, при которой отсутствуют все комиссуральные структуры вместе с фрагментами прозрачной перегородки, и частичную агенезию, которая в свою очередь подразделяется на агенезию ростральных и каудальных отделов мозолистого тела [29]. Чаще встречается агенезия каудальных отделов мозолистого тела, которая может сочетаться с изолированной дилатацией задних отделов боковых желудочков - кольпоцефалией. КТ-признаки агенезии мозолистого тела включают визуализацию межполушарной кисты, смещение вверх расширенного III желудочка и специфическое изменение формы тел боковых желудочков с увеличением расстояния между ними в виде симптома «ухвата» [7, 31]. Определяется расширение задних рогов боковых желудочков, своеобразный U-образный характер передних (фронтальных) рогов, удлиненная форма отверстия Монро. Кроме того, часто выявляются признаки атрофии вещества головного мозга в виде углубления и расширения конвекситальных борозд, расширения межполушарной и латеральной щелей, множественные пороки развития головного мозга.
S. Carney и соавт. [9] описали девочку с сочетанием признаков двух синдромов: Айкарди и де Морсье (септооптической дисплазии).
При записи паттерн-реверсивных зрительных вызванных потенциалов (ЗВП) у детей регистрируются ответы как на низко-, так и высокочастотные стимулы [1].
У детей раннего возраста с синдромом Айкарди, не имеющих нарушений со стороны оптических сред, макулы и диска зрительного нерва, острота зрения, определенная при помощи паттерн-реверсивных ЗВП или методом предпочтительного взора, может соответствовать возрастной норме [1, 19].
Синдром Айкарди необходимо дифференцировать от других мультисистемных заболеваний, характеризующихся церебральными аномалиями, изменениями диска зрительного нерва и сетчатки: туберозный склероз, септооптическая дисплазия, папилло-ренальный синдром, врожденные TORH-инфекции, перинатальные гипоксически-ишемические поражения, изолированная агенезия мозолистого тела, сочетанные и изолированные церебральные аномалии (полимикрогирия, гетеротопия, лиссэнцефалия) [1, 2, 9, 13].
В настоящее время лечение синдрома Айкарди носит исключительно паллиативный характер. Основная стратегия терапии - купирование эпилептических приступов у детей грудного возраста, т.е. инфантильных спазмов, которые зачастую резистентны к противосудорожной терапии. Применяют различные комбинации антиконвульсантов в максимально переносимых дозах. Для монотерапии применяют противоэпилептические препараты - вальпроат (конвулекс-капли) 30-100 мг/кг в сутки в 2-3 приема или вигабатрин (сабрил) 50-150 мг/кг в сутки (средняя суточная доза 100 мг/кг) в 2 приема. При неэффективности монотерапии переходят к политерапии: 1) вальпроат 30-80 мг/кг в сутки в 2-3 приема + вигабатрин (сабрил) 50-150 мг/кг в сутки в 2 приема; 2) вальпроат 30-80 мг/кг в сутки в 2-3 приема + этосуксимид 20-35 мг/кг в сутки в 2-3 приема; 3) вальпроат 30-80 мг/кг в сутки в 2-3 приема + фризиум 0,5-1,0 мг/кг в сутки (максимально 1 мг/кг в сутки) в 2 приема. Высокоэффективны кортикостероиды: синактен-депо, начиная с 0,1 мг внутримышечно 1 раз в сутки, с постепенным наращиванием по 0,1 мг 1 раз в 2-5 дней до дозы 1,0 мг в сутки. Продолжительность терапии синактеном-депо составляет 1-3 мес. Преднизолон (2-10 мг/кг в сутки), гидрокортизон (5-20 мг/кг в сутки) применяют перорально. Продолжительность лечения варьирует от 3 до 8 нед, возможна длительная терапия - 4-6 мес. Гормональную терапию целесообразно назначать в сочетании с базовыми антиконвульсантами [3].
Таким образом, практически у всех детей с синдромом Айкарди присутствует нарушение психомоторного развития различной степени. От возраста дебюта эпилептических приступов зависит степень тяжести когнитивного дефицита.
На ЭЭГ при инфантильных спазмах в ряде случаев регистрируются типичные изменения в виде гипсаритмии. Методы прижизненной нейровизуализации - НСГ, КТ, МРТ головного мозга - окончательно подтверждают агенезию мозолистого тела у детей с синдромом Айкарди. Хориоретинальные лакунарные очаги, увеличение экскавации диска зрительного нерва, сочетающиеся с поражениями постгеникулярных зрительных путей, в частности зрительной лучистости, определяемыми при радиологических исследованиях, являются офтальмоскопическими проявлениями данного заболевания.
Ранняя диагностика синдрома Айкарди, включающая невро-, радио- и офтальмологические исследования, позволяет достаточно полно представить диагностический профиль этого тяжелого и прогностически неблагоприятного заболевания.
Кафедра неврологии факультета усовершенствования врачей и кафедра нервных болезней педиатрического факультета Российского государственного медицинского университета, Москва
Тушинская городская детская больница, Москва
ГБОУ ДПО «Российская медицинская академия последипломного образования» Минздрава России, Москва, Россия
Современная диагностика агенезии мозолистого тела у детей
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2017;117(1): 63‑66
В статье представлены наблюдения авторов, касающиеся основных нейровизуализационных (МРТ) проявлений агенезии мозолистого тела (АМТ). Этому предпослан обзор литературы по строению МТ в норме и патологии. Подчеркивается, что часть случаев изолированной АМТ выявлена при рутинном пренатальном УЗИ. Пренатальная МРТ более эффективна в этом отношении. В 74% наблюдений у больных с АМТ результаты МРТ совпадают с данными УЗИ и КТ. МРТ обладает преимуществом в диагностике врожденных пороков развития МТ, а также дифференциации сопутствующих аномалий ЦНС.
Мозолистое тело (МТ) — самая крупная церебральная комиссура, располагающаяся по срединной продольной линии головного мозга. Оно соединяет филогенетически наиболее молодые участки больших полушарий мозга, обеспечивая межполушарную передачу информации [1].
Агенезия мозолистого тела (АМТ) — отсутствие пересечения средней линии комиссуральными волокнами М.Т. По классификации врожденных пороков развития головного мозга и черепа D. Harwood-Nash [2] АМТ относится к группе нарушений органогенеза и занимает 10-е место среди всех других пороков развития ЦНС. Частота АМТ составляет 0,3—0,7% в общем населении и 2—3% — среди инвалидов с умственной отсталостью [3].
В сочетании с АМТ могут встречаться такие пороки развития, как липомы МТ, полимикрогирия, шизэнцефалия, межполушарные кисты, мальформация Денди—Уокера и др. Как правило, АМТ не имеет самостоятельной клинической картины, что обусловливает сложности ее диагностики [4]. Прижизненная визуализация АМТ стала возможной благодаря внедрению в клиническую практику магнитно-резонансной томографии (МРТ) головного мозга.
Механизмы формирования МТ до настоящего времени остаются предметом дискуссий. Известно, что нервная система развивается из медуллярной трубки. На 4-й неделе гестации образуются три первичных мозговых пузыря, на 6—7-й неделе — пять. МТ образуется из первого мозгового пузыря и формируется из покровной части конечной пластинки Гиса, дорсальный конец которой утолщается, образуя так называемый поперечный вал [5]. Согласно результатам ряда исследований [5—7], на 15—17-й неделе внутриутробного развития появляются комиссуральные волокна МТ, соединяющие большие полушария мозга. По данным G. Davilla-Gutierrez [8], вначале МТ увеличивается рострально, затем каудально, при этом волокна ростральной части пересекаются приблизительно на 74-й день, а на 115-й день происходит его утолщение. L. Richards и соавт. [7], напротив, указывают, что часть каудальных аксонов МТ, зависящих от гиппокампа, может одновременно пересекаться с ростральными аксонами МТ. К 20-й неделе МТ практически сформировано [9]. У недоношенных детей его формирование замедляется [10]. У младенцев к 8—10-му месяцу жизни происходит окончательная миелинизация ростральной части МТ [11].
АМТ может быть результатом недостаточного развития комиссуральной пластинки, агенезии или деструкции третьего слоя нейронов [12]. Есть данные, подтверждающие, что первичная АМТ формируется до 12—16-й недели внутриутробного развития [13]. После 18—20-й недели происходит вторичное повреждение МТ, являющееся следствием энцефаломаляции на более поздних этапах развития плода [14]. При вторичной АМТ чаще происходит недоразвитие задней части (валик) МТ, а передняя или средняя части МТ (колено, клюв и часть корпуса) отсутствуют [12].
Определенные трудности в диагностике АМТ сохраняются и в наши дни. Время ее внутриутробного обнаружения напрямую зависит от стадии онтогенеза МТ.
По данным R. Achiron и соавт. [15], которые обследовали 270 беременных между 16-й и 37-й неделями гестации, при ультразвуковом исследовании (УЗИ) плода удалось измерить длину, ширину и толщину МТ в 258 случаях. Это позволило выявить линейную зависимость между увеличением роста МТ и гестационным возрастом плода, а также установить, что на 19—21-й неделях гестации происходит его максимальное увеличение. A. Barkovich и соавт. [16] предположили, что определенные подтипы АМТ зависят от пола. Это нашло подтверждение в экспериментах на животных: у лабораторных животных (мыши) линии BALB/cCF с нарушениями развития МТ было выявлено преобладание самок [17].
Согласно исследованиям P. Govaert и L. de Vries [11], после 20-й недели беременности на УЗИ определяются следующие признаки АМТ: вентрикуломегалия; высокое расположение III желудочка; отсутствие полости прозрачной перегородки, а по данным С.М. Воеводина [18], на УЗИ плода при условии технической чистоты получения сагиттальной проекции АМТ выявляется на сроках 15—36 нед беременности. К сожалению, значительная доля случаев изолированной АМТ при рутинном пренатальном УЗИ не улавливается.
Пренатальная МРТ наиболее эффективна при диагностике АМТ [11]. В 74% наблюдений у больных с АМТ результаты МРТ совпадают с данными УЗИ и КТ-диагностики [19].
Постнатальная ультразвуковая картина АМТ характеризуется отсутствием изображения МТ; исчезновением нормальной архитектоники борозд и извилин в сагиттальной плоскости сканирования; веерообразным отхождением борозд от крыши III желудочка; широким расположением боковых желудочков с изменением ориентации передних рогов в коронарных плоскостях и смещением вверх или расширением III желудочка. На К.Т. определяются параллельный ход и увеличение расстояния между телами боковых желудочков, расширение задних рогов и преддверий боковых желудочков (кольпоцефалия) [11].
В данной публикации приведены собственные наблюдения АМТ и анализ соответствующих данных прижизненной нейровизуализации мозга.
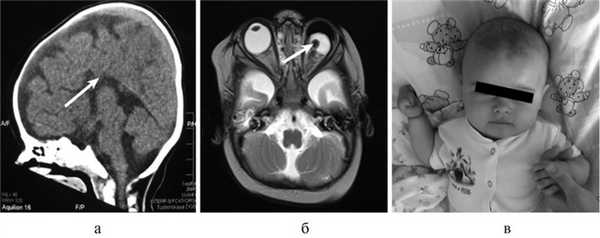
Рис. 1. КТ и МРТ головного мозга (сагиттальная, фронтальная проекции) и внешний вид больной Т., 8 мес. а — КТ: АМТ (стрелка); б — микрофтальм слева на МРТ (стрелка); в — внешний вид пациентки с множественными стигмами дизэмбриогенеза.
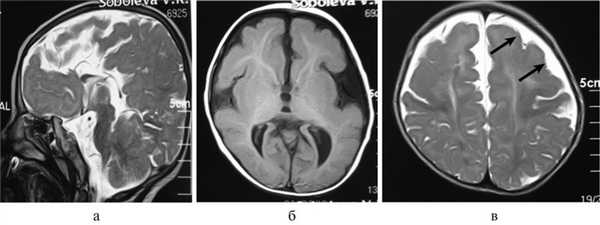
Рис. 2. Результаты обследования больной С., 8 лет. а, б, в — МРТ головного мозга (сагиттальная и аксиальная проекции): сочетание АМТ и диффузной пахигирии (стрелки).
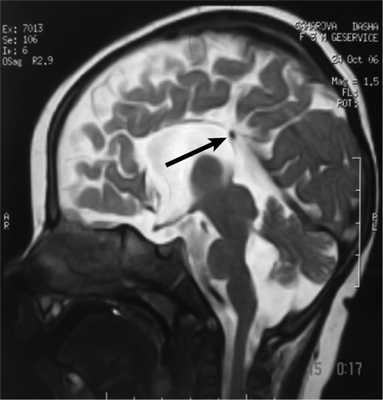
Рис. 3. МРТ головного мозга (сагиттальная проекция) больной С., 7 лет. АМТ (стрелка).
Важной находкой при АМТ на МРТ является параллельная ориентация тел боковых желудочков по отношению друг к другу (рис. 4, а), расширенные фронтальные отделы желудочков, так называемый «симптом ухвата».
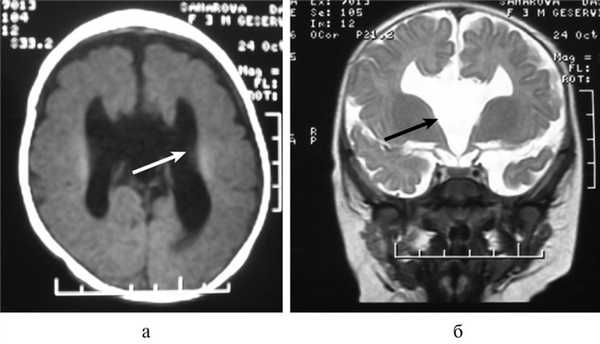
Рис. 4. МРТ головного мозга больной К., 4 года. АМТ. Аксиальная проекция: а — аномальная параллельная ориентация тел боковых желудочков по отношению друг к другу (стрелка); б — фронтальное сечение: деформация передних и задних рогов боковых желудочков, своеобразный U-образный характер фронтальных отделов боковых желудочков (стрелка).
При МРТ мы обращали внимание также на описанное в литературе [12] изолированное расширение задних рогов — кольпоцефалию, обусловленную гипоплазией ассоциативных трактов белого вещества затылочных долей. Диагностический ряд на МРТ дополняют отсутствие нормально сформированных перикаллезных извилин (рис. 5, а) и радиальная центростремительная позиция борозд медиальной поверхности теменной доли (рис. 5, б).
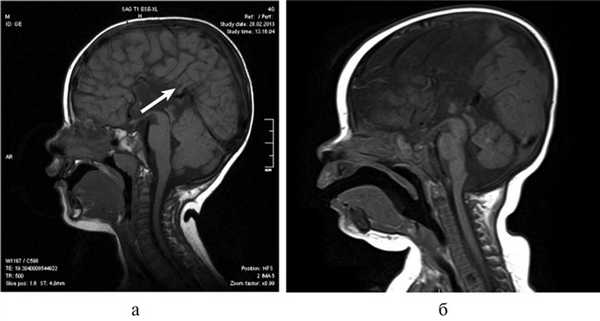
Рис. 5. МРТ головного мозга больного А., 3 года. АМТ. а — сагиттальная проекция. Отсутствие нормально сформированных перикаллезных извилин (стрелка), б — высокое положение III желудочка, радиальная центростремительная направленность борозд медиальной поверхности теменных долей.
Появилась также новая нейровизуализационная технология — магнито-резонансная морфометрия, которая помогает оценить степень поражения М.Т. Можно вычислить каллозальный индекс — отношение площади МТ к площади внутреннего сечения черепной коробки, оценить размер, общую площадь, толщину колена, корпуса, перешейка и валика МТ [22—24].
Нередко изменения структуры МТ обнаруживаются случайно, при КТ- или МРТ-исследованиях по поводу других заболеваний. Так, описано [25] наличие АМТ у пациента с ювенильной болезнью Гентингтона в сочетании с множественными соединительнотканными дисплазиями, остеохондропатией.
Таким образом, среди множественных врожденных пороков развития ЦНС, которыми страдают около 1% детей [26], одно из ведущих мест принадлежит АМТ. В настоящее время совершенствуются методы нейровизуализации, такие как МРТ плода, применяются методики ультразвукового сканирования плода во внутриутробном периоде на ранних сроках гестационного развития, позволяющие заподозрить тот ли иной наследственный синдром в пренатальном периоде.
Адекватное и своевременное распознавание врожденных пороков развития МТ представляет собой непростую задачу, решение которой требует владения новейшими методиками нейровизуализационной диагностики. В данной статье представлены основные нейровизуализационные проявления АМТ.
Синдром Айкарди
Синдром Айкарди — это Х-сцепленное генетическое заболевание, характеризующееся сочетанием агенезии мозолистого тела с формированием хориоретинальных лакун и вариабельными эмбриональными аномалиями скелета. Типичной клинической картиной выступает триада признаков: инфантильные спазмы, задержка психического развития, нарушения зрения. Диагностика осуществляется при помощи электроэнцефалографии, церебральной МРТ, офтальмоскопии, изучения зрительных потенциалов, рентгенографии. Генетические методы диагностики пока не найдены. Лечение паллиативное с назначением комбинаций антиконвульсантов для постоянного приема, периодических курсов кортикостероидов.
МКБ-10



Общие сведения
Синдром Айкарди относится к редким расстройствам эмбрионального развития нервной системы. Впервые описан в 1965 году французским детским неврологом Жаном Айкарди как заболевание, характеризующееся следующей триадой признаков: агенезия мозолистого тела, офтальмологические расстройства, мышечные спазмы. Название синдрому в честь Ж. Айкарди было дано в 1972 г. Дальнейшее его изучение расширило список характерных клинических признаков, включив в них лицевой дисморфизм, умственную отсталость, аномалии позвоночного столба. Развитие методов нейровизуализации и генетических исследований позволило сделать диагностику более точной. По данным мировой статистики, всего насчитывается около 500 заболевших. Синдром связан с патологией Х-хромосомы, встречается исключительно у девочек. У плодов мужского пола возникают аномалии, не совместимые с жизнью.

Причины
Заболевание является генетической патологией, связанной с нарушением участка на Х-хромосоме. Считается, что оно имеет доминантное сцепленное с Х-хромосомой наследование. Теоретически вероятность рождения больной девочки от матери, страдающей синдромом, составляет 50%. Наличие патологической Х-хромосомы у мальчиков приводит к внутриутробной смерти.
На практике генетическая передача не зафиксирована, поскольку заболевшие девочки не могут иметь потомства в силу выраженных психомоторных расстройств. Все исследованные случаи синдрома Айкарди возникали de novo. Встречаются преимущественно спорадические варианты заболевания. Исключение составляет единственный известный случай у двух сестер. В связи с малым количеством заболевших, проведение достоверного исследования мутагенных триггеров затруднительно.
Патогенез
Патогенетические механизмы развития заболевания находятся в стадии изучения. Предполагается, что мутации генов обуславливают расстройство нейрональной миграции, происходящей в период между 12-й и 24-й неделями эмбрионального развития. В эмбриогенезе миграция неокортикальных нейронов на периферию приводит к образованию коры и подкорковых зон. Ее нарушения обуславливают дисгенезию мозолистого тела, аномальное формирование извилин и архитектоники мозга.
В норме в мозолистом теле проходят межполушарные проводящие пути, поэтому его дисэмбриогенез влечет нарушение связей между полушариями. Макроскопически в головном мозге выявляется микрогирия, субатрофия церебральной коры, перивентрикулярные кисты, гетеротопия, агенезия мозолистого тела. Микроскопически в сетчатке отмечается истончение слоев, уменьшение количества сосудов, гиперплазия пигментного эпителия.
Симптомы
Дети с синдромом Айкарди рождаются без видимых патологий, обычно в срок. В большинстве случаев беременность и роды протекают без осложнений. Заболевание манифестирует инфантильными спазмами в первые месяцы жизни. Спазмы представляют собой фокальные эпиприступы, при патологии Айкарди отличаются большой вариабельностью.
Возможны миоклонии, тонические судороги, ретропульсии, пропульсии. Наблюдаются симметричные и асимметричные, единичные и серийные судорожные приступы, типична тенденция к серийности. Флексорные спазмы отмечаются у 97% пациенток. В 42% случаях заболевания инфантильные спазмы сочетаются с другими видами эпиприступов, как фокальными, так и генерализованными. Фокальные припадки происходят с вовлечением преимущественно лицевой мускулатуры.
Задержка психомоторного развития характерна для всех больных синдромом. Отмечается обедненность эмоциональных реакций, утрачен контакт с окружающими, останавливается развитие навыков и предречевое развитие. Позже присоединяются моторные расстройства: геми- и тетрапарезы с повышением или снижением тонуса мышц, возможна спастичность. В отдельных случаях описана умеренная задержка развития, легкая степень олигофрении.
Офтальмологические аномалии вариабельны, включают: атрофию зрительного нерва, микроофтальмию, колобому. Возможна катаракта, пигментный ретинит. Клинически наблюдается значительное снижение зрительной функции. Со стороны желудочно-кишечного тракта характерна неустойчивость стула, проблемы с кормлением, гастроэзофагальный рефлюкс.
Среди челюстно-лицевых дисморфий описаны асимметрия лица, гипертелоризм, аномалии ушной раковины, выступающие резцы. У 33% пациенток отмечаются дисэмбриогенетические изменения позвоночника и ребер в виде полупозвонков, расщепления позвоночника, агенезии ребра, аномального реберно-позвоночного сочленения. В возрасте после 7 лет типично замедление роста, в пубертате — задержка полового развития.
Гемангиомы, невусы и другие дерматологические проявления описаны у 20% больных. В 7% случаев синдром Айкарди протекает с аномалиями конечностей: гипоплазией пальцев, синдактилией, каптодактилией.
Осложнения
Выраженная олигофрения, двигательные расстройства с первых месяцев жизни обуславливают глубокую инвалидность ребенка. Девочки нуждаются в постоянном уходе, половина из них неспособны к элементарному самообслуживанию. Нарушение мышечной иннервации при отсутствии постоянных реабилитационных мероприятий сопровождается гипотрофиями, в случаях спастичности — развитием контрактур.
Судорожный синдром с генерализованными припадками и кластерными приступами усугубляет неврологический дефицит, олигофрению. Позвоночные аномалии приводят к развитию выраженного сколиоза. Больные с синдромом Айкарди подвержены рецидивирующей пневмонии, имеют больший риск возникновения новообразований.
Диагностика
Внешний вид ребенка с лицевыми аномалиями, уменьшенным кончиком носа, латерально расположенными бровями позволяет врачу предположить генетическое заболевание. В неврологическом статусе показательны мышечная гипотония или односторонняя спастичность, снижение мышечной силы в конечностях, оживление рефлексов, снижение психического развития. Диагностика синдрома осуществляется совместно с генетиками, однако специфический генетический анализ еще не разработан. Назначаются следующие исследования:
- Электроэнцефалография. Наиболее типичным ЭЭГ-признаком синдрома Айкарди выступает гипсаритмия, появляющаяся после манифестиции инфантильных спазмов. Типично различие ЭЭГ-паттерна двух полушарий, феномен «расщепленного мозга». У ряда пациенток энцефалографическая картина имеет стертый характер. Наличие вариабельных судорожных припадков обуславливает полиморфизм изменений ЭЭГ.
- Офтальмоскопия. Выявляет наличие хориоретинальных лакун, визуализирующихся как белесоватые депегментированные участки сетчатки. Двусторонние лакуны расположены асимметрично, в 10-20% случаев очаги диагностируются только в одном глазу. При офтальмоскопии пациенток более старшего возраста диагностируется катаракта, колобома и/или атрофия диска зрительного нерва.
- Зрительные вызванные потенциалы (ЗВП). Исследуются для определения остроты зрения в младенческом возрасте и проведения дифдиагностики с другими офтальмологическими заболеваниями. У маленьких пациенток без изменений на глазном дне острота зрения может соответствовать возрастной норме.
- МРТ головного мозга. Визуализирует полное или частичное недоразвитие мозолистого тела, признаки корковой атрофии, пахигирию, расширение борозд. III желудочек расширен, вместе с боковыми желудочками смещен вверх, вокруг него выявляются церебральные кисты. Отмечается изменение формы латеральных желудочков, множественные области гетеротопии с отсутствием дифференцировки слоев коры, гидроцефалия.
- Рентгенография скелета. Необходима для диагностики костных аномалий, сопровождающих синдром Айкарди.
Перинатальная ДНК-диагностика не разработана. Выявление патологии возможно в ходе ультразвукового исследования беременной. По данным УЗИ 1 триместра возможна диагностика агенезии мозолистого тела. Позже выявляются микроцефалия, интракраниальные кисти, позвоночные аномалии, прочие пороки развития.
Дифференциальная диагностика
Необходимо дифференцировать патологию Айкарди от прочих ранних мультисистемных поражений, включающих сочетание дисгенеза церебральных структур и нарушения строения сетчатки. Первым симптомом септооптической дисплазии выступает горизонтальный нистагм, отсутствующий у пациентов с синдромом Айкарди. Отличительной чертой туберозного склероза служит наличие дерматологических симптомов: гиперпигментаций, ангиофибром, участков «шагреневой» кожи. Папилло-ренальный синдром протекает с поражением почек. Дифференциальная диагностика с врожденными TORH-инфекциями проводится на основании данных лабораторных обследований.
Следует отличать синдром Айкарди от заболевания Айкарди-Гутиера с аутосомно-рецессивным типом наследования. Последнее относится к прогрессирующим лейкодистрофиям детского возраста, характеризуется потерей приобретенных навыков, лимфоцитозом цереброспинальной жидкости, повышенным уровнем альфа-интерферона, кальцификатами в подкорковых структурах.
Лечение синдрома Айкарди
В основе терапии заболевания лежит паллиативная медикаментозная помощь и реабилитационные мероприятия. Базовой схемой является сочетание противосудорожных и гормональных фармпрепаратов, что позволяет добиться снижения частоты эпиприступов. Основными компонентами лечения являются:
- Антиконвульсанты. На первом этапе проводится монотерапия в максимально переносимой дозировке. Поскольку инфантильные спазмы при синдроме Айкарди отличаются стойкостью к противосудорожным препаратам, то приходится переходить к комбинированной схеме терапии, включающей 2-3 препарата.
- Глюкокортикоиды. Показали свою эффективность при внутримышечном введении в течение 1-3 месяцев. В ряде случаев по назначению врача возможна более длительное глюкокортикоидное лечение.
- Реабилитационная терапия. Направлена на поддержание двигательной активности, предотвращение мышечной атрофии, формирования контрактур при спастичности. Лечебные мероприятия включают массаж и специальную гимнастику, разработанные индивидуально с учетом имеющихся аномалий. Детям со средней степенью когнитивного дефицита показаны занятия с психологом, логопедом для максимально возможного развития навыков.
Прогноз и профилактика
Поскольку синдром Айкарди является генетическим заболеванием с неуточненными этиофакторами генных мутаций, то его специфическая профилактика затруднительна. Возможно проведение общих профилактических мероприятий, направленных на охрану женщины от вредоносных воздействий в период беременности.
1. Синдром Айкарди/ Милованова О.А.// Журнал неврологии и психиатрии им. С.С. Корсакова - 2011. - 111(7).
2. Синдром Айкарди/ Петухова Н.М., Павловец Л.П., Жакашева А.С.// Вестник Алматинского государственного института усовершенствования врачей. - 2010. - 1.
3. Aicardi syndrome: an epidemiologic and clinical study in Norway/ Lund, C., Bjørnvold, M., Tuft, M., Kostov, H., Rosby, O., Selmer, K. K.// Pediatric Neurology. - 2015. - 52(2).
4. Phenotype and management of Aicardi syndrome: new findings from a survey of 69 children/ Glasmacher, M. A., Sutton, V. R., Hopkins, B., Eble, T., Lewis, R. A., Park Parsons, D., & Van den Veyver, I. B. // Journal of child neurology. - 2007. - 22(2).
Синдром ЕЕС. Возможности пренатальной диагностики и особенности медико-генетического консультирования
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Введение
Синдром ЕЕС (ectrodactyly-ectodermal dysplasia-clefting syndrome) - редкий генетический синдром, обычно проявляющийся триадой признаков: расщелиной лица, эктродактилией конечностей и признаками эктодермальной дисплазии [1]. Выделяют 2 типа синдрома: ЕЕС-1 и ЕЕС-3. Ранее был описан также и ЕЕС-2, однако на сегодняшний день локализация гена, кодирующего ЕЕС-2, считается ошибочной, следовательно, и формы ЕЕС-2 не существует. Первый тип характеризуется мутацией в 7-й хромосоме в области 7q11.2-q21.3, третий - имеет в большинстве случаев мутацию Тp63-гена и другие новые, спонтанные мутации на 3-й хромосоме 3q27 [2]. В OMIM (ежедневно обновляемом международном классификаторе Online Mendelian Inheritance in Man), где представлены все известные на сегодняшний день фенотипы и генотипы менделирующих (наследуемых) болезней человека, эти виды синдрома кодируются по-разному: ЕЕС-1 - 129900, а ЕЕС-3 - 604292 [3].
ЕЕС-синдром - наследственная патология с аутосомно-доминантным типом наследования, характеризующаяся различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности) [1].
Манифестные признаки синдрома ЕЕС (синдромальное ядро)
Эктродактилия - это расщелина кистей и/или стоп с отсутствием одного или нескольких центральных пальцев, известная также под названием "Split hand-split foot malformation" (SHFM). Часто внешний вид кистей и стоп сравнивают с клешней краба. Нередко встречается в сочетании с синдактилией (сращением пальцев).
Эктодермальная дисплазия проявляется особенностями строения всех производных эктодермы. Изменения затрагивают волосы, зубы, ногти [11]. Волосы и ресницы у таких людей тусклые, редкие и жесткие. Характерны аномалии зубов, гипоплазия зубной эмали, снижение пигментации волос, кожи, радужной оболочки глаз, что вызывает светлый их цвет. Также часто сочетание синдрома с фотобоязнью [1], обструкцией носо-слезных каналов [12], кондуктив- ной тугоухостью [13].
Эктодермальная дисплазия проявляется отсутствием потовых желез, что характеризуется наличием гипогидротинового синдрома, проявляющегося в резкой сухости и шелушении кожи. Характерным проявлением этого синдрома является хриплый, осипший голос из-за нарушения увлажнения голосовых связок, вследствие чего не происходит полного их смыкания [14].
Нарушение речи у больных с ЕЕС-синдромом имеет много причин. Во-первых, это связано с последствиями наличия расщелины губы и/или неба и "гиперназальной" речью, во-вторых, правильной артикуляции может препятствовать не только изменение голосовых связок, но и патология зубов. В-третьих, из-за аномалий слуховых косточек часто прогрессирует тугоухость, ведущая к когнитивным расстройствам и расстройствам речи.
Дети с этим синдромом обычно имеют нормальный уровень интеллекта. Лечение основано на коррекции пороков лица и конечностей. Признаки эктодермальной дисплазии проявляются в постнатальном периоде [1]. Врожденные пороки развития (ВПР) мочевыделительной системы у больных с синдромом ЕЕС возникают более чем в половине случаев. Среди них описаны: мегауретер, гидронефроз, уретероцеле, аплазия почки, аномалии гениталий, уретровезикальный рефлюкс с частыми инфекциями мочевыделительных путей [12].
Пренатальная диагностика синдрома ЕЕС
Учитывая выраженность проявлений при синдроме ЕЕС, пренатальная диагностика этой патологии, безусловно, возможна. Однако на сегодняшний день в мировой литературе встречается незначительное число публикаций, посвященных диагностике этого редкого синдрома 16, что связано, видимо, с редкостью возникновения данной аномалии. В основном сроками постановки диагноза являются 16-30 недель беременности. Большую помощь в диагностике данной патологии, учитывая выраженные изменения фенотипа лица и конечностей, оказывает применение новых ультразвуковых технологий 3D/4D с методиками поверхностной реконструкции [16].
Первое описание пренатальной диагностики синдрома датировано 1993 годом и принадлежит M. Bronshtein и R. Gershoni, когда при применении трансвагинальной эхографии у плода была выявлена расщелина губы и неба и эктродактилия в 14 недель беременности [17]. Уникальность этого факта в том, что, пожалуй, впервые из известных случаев диагностики врожденных аномалий дебют дородового выявления порока принадлежит сроку первого триместра. Примечательно и то, что авторами не только описаны отдельно выявленные пороки развития плода, но и пренатально установлена нозология этого состояния. Другими словами, диагноз не звучал, как "множественные врожденные пороки развития", когда невозможно говорить об этиологии заболевания, а следовательно, и о мерах специфической профилактики данной патологии в дальнейшем в семье. Пренатальный диагноз был выставлен полно и абсолютно корректно, что особенно важно при проведении медико-генетического консультирования с формированием тактики адекватного репродуктивного поведения семьи в дальнейшем.
Из-за незначительного количества публикаций, посвященных пренатальной диагностике этого генетического синдрома, представляем ряд собственных наблюдений диагностики синдрома ЕЕС в разные сроки беременности, в том числе и в первом триместре. Особо рассмотрим особенности проведения медико-генетического консультирования при диагностике различных форм синдрома с аутосомно-доминантным типом наследования для выработки специфических мер профилактики данного наследственного заболевания.
Клиническое наблюдение 1
Пациентка Н. 24 лет. Беременность первая. Женщина и муж соматически здоровы, брак неродственный. Обратилась в медико-генетическое отделение МОНИИАГ в 21,4 недели беременности в связи с подозрением на порок развития конечностей у плода. Были выявлены: эктродактилия кистей (рис. 1) и стоп плода (рис. 2.). Из особенностей строения выявлены двусторонние пиелоэктазии (рис. 3). Проведено медико-генетическое консультирование. Семья приняла решение прервать данную беременность в связи с наличием инвалидизирующих пороков конечностей. При патологоанатомическом исследовании ВПР конечностей подтверждены, а также дополнительно выявлена расщелина мягкого неба и признаки эктодермальной дисплазии (гипертрофия десен, характерный лицевой фенотип). Окончательный диагноз: "синдром ЕЕС, аутосомно-доминантный тип наследования". Мутация de novo.
Читайте также: