УЗИ, МРТ при синдроме Гольденхара у плода
Добавил пользователь Skiper Обновлено: 22.01.2026
На приём к генетику явилась семейная пара с ребёнком (1 месяц) с жалобами на отсутствие левой ушной раковины, левого слухового прохода, наличие папилломподобных выростов на левой щеке, деформацию рта.
Жалобы
У ребенка с рождения отсутствовала левая ушная раковина, левый наружный слуховой проход, на левой щеке присутствовали 3 преаурикулярных выроста, деформация рта (микростомия, левосторонняя расщелина губы).
Анамнез
Ребенок от второй беременности, вторых срочных родов. Беременность протекала с угрозой в первом триместре, на сроке семи недель женщина перенесла ветряную оспу с подъёмом температуры. Вес ребёнка при рождении — 3400 гр, рост — 53 см, по шкале Апгар — 8/9 баллов. При рождении была замечена аплазия левой ушной раковины, отсутствие наружного слухового прохода, преаурикулярные выросты, расщелина губы. Выписаны из роддома на четвёртые сутки с диагнозом "Множественные врождённые пороки развития. Неонатальная желтуха".
Обследование
Ребёнок женского пола, возраст — 1 месяц. Вес — 3900 гр, рост — 55 см. При осмотре обнаружено, что у ребёнка уменьшена левая половина лицевого черепа (гемифациальная гипоплазия), отсутствует левая ушная раковина, на её месте имеется папиллярный вырост, ещё два выроста локализуются на левой щеке. Левый наружный слуховой проход не визуализируется. Микростомия, расщелина верхней губы, протяженностью 4 мм, справа. При осмотре ротовой полости обнаружено уменьшение левого альвеолярного отростка верхней челюсти. Физическое развитие в пределах нормы. Отклонений со стороны дыхательной, пищеварительной, мочевыделительной, половой систем не выявлено.
Кариотипирование: 46ХХ (нормальный женский), скрининг на наследственные болезни обмена — норма. Аудиограмма справа — норма, слева — слух отсутствует. Общий анализ крови: гипохромная микроцитарная анемия лёгкой степени. После проведённой коррекции анемии общий анализ крови без патологии. Общий анализ мочи: без патологии. Биохимические исследования крови: без патологии.
Диагноз
Лечение
В возрасте двух лет хирургически были удалены папиллярные выросты. С возрастом будет возможна косметическая коррекция расщелины губы.Возможна косметическая коррекция ушной раковины при помощи импланта.
Коррекция косметических недостатков проводится в разном возрасте, так как она не несёт в себе опасности для жизни и развития. На удалении преаурикулярных выростов настояли родители. Вопрос о слухопротезировании будет решаться в более старшем возрасте, учитывая, что слух справа в норме. После прорезывания зубов показана ортодонтическая коррекция верхней челюсти.
Благодаря хирургическому лечению удалось добиться коррекции косметических дефектов. Ребёнок развивается по возрасту, речь в пределах возрастной нормы.
Заключение
Прогноз при синдроме Гольденхара благоприятный, лечение заключается в косметической коррекции, слухопротезировании при возможности. Данное заболевание не наследуется. Существуют данные, что развитие синдрома Гольденхара связано с болезнью матери во время беременности на малых сроках, описаны случаи, связанные с острыми респираторными заболеваниями, ветряной оспой, гриппом. Возможна пренатальная диагностика заболевания при помощи ультразвукового исследования. При подозрении на синдром Гольденхара обязательно проводится исследование кариотипа для исключения фенокопий. Патогенез заболевания связан с нарушением формирования первой и второй жаберных дуг в процессе эмбриогенеза под воздействием, предположительно, внешних факторов.
Синдром Гольденхара ( Окуло-аурикуло-вертебральная дисплазия )
Синдром Гольденхара — это редкое врожденное заболевание, которое проявляется множественными пороками развития, выраженным клиническим полиморфизмом. Возникает вследствие мутации генов, локализованных в 5, 14, 20 хромосомах. Для синдрома характерны разнообразные аномалии лицевого скелета, патологии органов чувств, зачастую болезнь сопровождается задержкой психического развития. План обследования включает генетическое тестирование для верификации диагноза, лабораторно-инструментальные методы с учетом ведущих клинических признаков. Лечение поддерживающее, многим больным требуется слухопротезирование, комплексная нейрореабилитация.
МКБ-10
Общие сведения
Синдром назван в честь американского ученого Мориса Гольденхара, который описал его типичные клинические проявления в 1952 г. В 1963 г. Р.Дж. Горлин и его коллеги сообщили о собственных наблюдениях пациентов, дав болезни второе название «окуло-аурикуло-вертебральная дисплазия». Состояние выявляется с частотой от 1:3500 до 1:7000 живорожденных новорожденных, соотношение мальчиков и девочек составляет 3:2. Если в семье есть больной ребенок, то вероятность появления синдрома Гольденхара у следующих детей — не более 3%.

Причины
Заболевание возникает при нарушении дифференцировки 1-2-й жаберных дуг, что провоцируется мутацией в генах GSC и TCOF1. В 98% случаев синдрома роль наследственности не удается проследить. Однако около 2% больных имеют родственников с аналогичной клинической симптоматикой. В литературе описаны случаи аутосомно-доминантного и аутосомно-рецессивного наследования.
Основными предрасполагающими факторами считаются:
- воздействие тератогенных факторов на ранних сроках гестации;
- наличие у матери сахарного диабета, избыточного веса;
- предшествующие искусственные прерывания беременности и выкидыши.
Патогенез
Как следствие, лобный, нижне- и верхнечелюстные эктодермальные отростки, происходящие из первой жаберной дуги, и ушные раковины, образованные 1 и 2 жаберными дугами, развиваются асимметрично. После рождения ребенка это проявляется специфическими изменениями верхней и нижней челюстей, глаз и глазниц, мимической и жевательной мускулатуры, структур наружного и среднего уха, аномалиями прикуса, дефицитом мягких тканей.
Симптомы
Основные признаки синдрома Гольденхара — аномалии строения лица, которые в 70% случаев являются правосторонними. У всех пациентов наблюдается асимметрия, недоразвитие нижней челюсти, уменьшение в размерах, деформация или отсутствие ушных раковин. Патологии органа слуха чаще бывают односторонними, сопровождаются атрезией слухового прохода, преаурикулярными выростами. Иногда формируется дополнительная рудиментарная ушная раковина.
Также характерно уменьшение размера глазных яблок (микрофтальмия), косоглазие, атрезия радужки, катаракта. Около 50% пациентов с болезнью Гольденхара имеют высокое готическое небо, широкий рот (макростомию), расщепление языка, аномальный прикус, отсутствие части зубов. В 40% случаев отмечаются косолапость, аномалии позвоночника (сколиоз, spina bifida), искривление ребер. У 30% больных возникают врожденные патологии внутренних органов: пороки сердца, гипоплазия легких, дисплазия почек.
Осложнения
Самым опасным последствием синдрома Гольденхара считается умственная отсталость, которая обусловлена двусторонней тугоухостью, снижением остроты зрения. При этом у большинства детей наблюдается нормальное функционирование нервной системы, однако сенсорная депривация создает затруднения при речевом и психомоторном развитии. Осложненное течение синдрома встречается при тяжелых врожденных пороках сердца, легких, почек.
Диагностика
В большинстве случаев предварительный диагноз устанавливается на основании характерных фенотипических признаков: деформации ушей, асимметрии лица, патологии развития нижней челюсти. Для подтверждения синдрома Гольденхара обязательно назначается консультация генетика, проводится комплекс диагностических мероприятий:
- Генетический анализ. Тестирование на специфическую мутацию генов 14q32, 5p15, MYT1 позволяет 100% подтвердить диагноз. Исследование выполняется в специальных генетических лабораториях с использованием методов секвенирования экзона, флуоресцентной гибридизации.
- Аудиометрия. Оценка слуха в динамике необходима всем пациентам, чтобы вовремя выявить кондуктивную тугоухость, обеспечить мероприятия по ее коррекции. Расширенное обследование у отоларинголога может включать импедансометрию, оценку рефлекса стапедиальной мышцы.
- Неврологический осмотр. Консультация детского невролога требуется для определения причин задержки психомоторного развития, оценки функционирования центральной и периферической нервной системы. При наличии показаний больного направляют на дообследование к психиатру.
- Инструментальная визуализация. Чтобы подтвердить или исключить соматические пороки, при синдроме Гольденхара проводятся рентгенография грудной клетки, эхокардиография, УЗИ органов брюшной полости. Для исследования ЦНС применяется КТ или МРТ головного мозга.
Лечение синдрома Гольденхара
Пациенты, у которых диагностирована болезнь Гольденхара, подлежат пожизненному динамическому наблюдению. Им назначаются регулярные обследования у офтальмолога, отоларинголога, невролога и других специалистов, чтобы контролировать состояние здоровья. Курсы поддерживающего лечения включают несколько направлений помощи, основными из которых являются:
- Метаболическая терапия. Для правильного развития ЦНС, улучшения когнитивных навыков используются ноотропы, витамины группы В.
- Нейрореабилитация. Чтобы улучшить речевое развитие, рекомендованы продолжительные курсы занятий с логопедами, сурдологами. По показаниям проводится психологическая коррекция.
- Специальное обучение. Пациентам с признаками ЗПР необходимо продолжать учебу в коррекционных классах с дефектологами, сурдопедагогами.
Прогноз и профилактика
При отсутствии у больного врожденных соматических пороков и достаточном уровне интеллекта прогноз благоприятный, большинство пациентов доживают до зрелого и пожилого возраста. Вызывают опасения случаи синдрома Гольденхара, которые сопровождаются кардиологическими, пульмонологическими или нефрологическими пороками, также неблагоприятный прогноз устанавливают при тяжелой умственной отсталости.
Основу профилактики синдрома составляет антенатальная охрана плода. Критическим периодом является первый триместр, поскольку именно в это время возникают характерные костные аномалии. Беременным женщинам рекомендовано избегать контакта с химикатами, рентгеновским излучением, а также соблюдать противоэпидемические меры для защиты от вирусных инфекций.
1. Клинический случай синдрома Гольденхара в психиатрической практике/ А.В. Ковалева// Acta Biomedica Scientifica. — 2020. — №3.
2. Клинический случай синдрома Гольденхара у новорожденного/ Л.Г. Киселева, Л.П. Мокеева, Ю.С. Тишкова, Н.В. Павловский// Вятский медицинский вестник. — 2015. — №46.
3. Диагностика синдрома Гольденхара в периоде новорожденности/ С.И. Мизинцева// Бюллетень Северного государственного медицинского университета. — 2012.
4. Особенности общеклинических проявлений синдрома Гольденхара/ И.А. Карякина// Системная интеграция в здравоохранении. — 2010. — №2.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
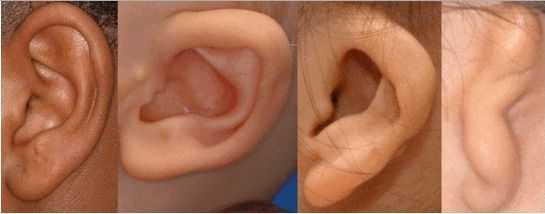
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
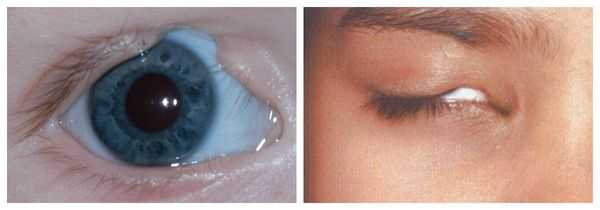
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
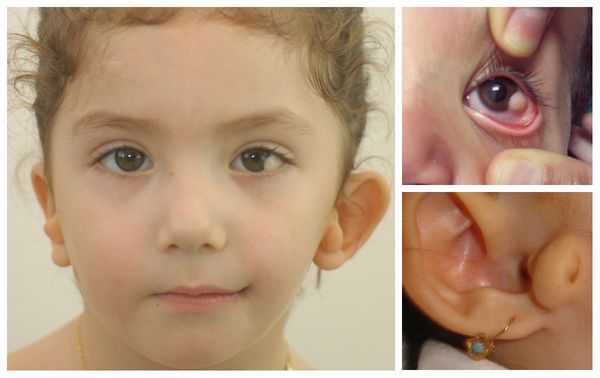
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
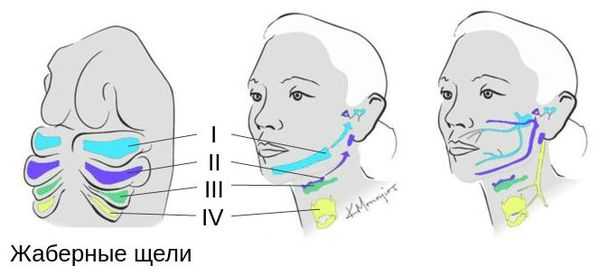
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
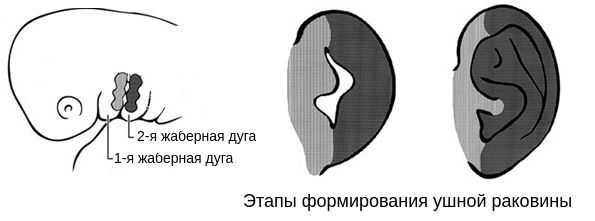
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
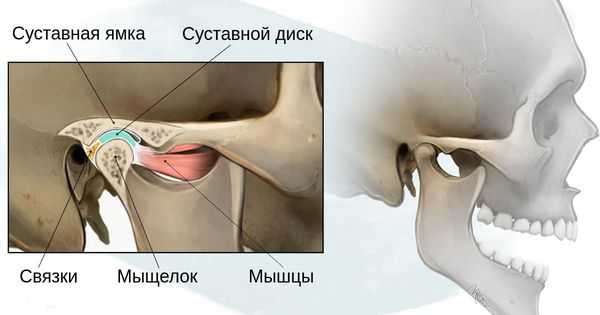
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
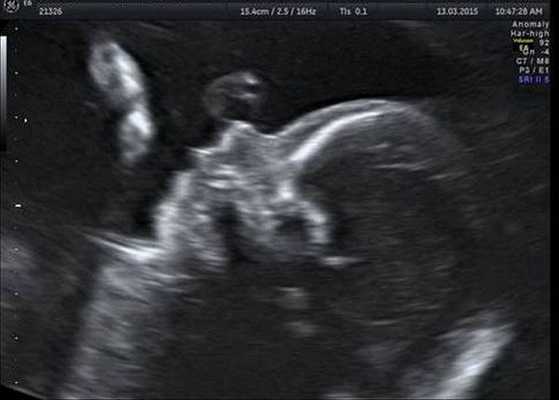
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Аномалии и патологии органов пищеварительной системы плода, выявляемые на УЗИ
Патологии органов пищеварительной системы встречаются у плода нередко как самостоятельно, так и в комплексе с другими аномалиями внутренних органов. На них приходится до 21% пороков у новорождённых и 34% случаев младенческой смертности.
Причины нарушений формирования органов ЖКТ у плода, статистика
Аномалии строения органов ЖКТ связаны с нарушением эмбриогенеза на стадии 4-8 недель беременности, когда идёт образование отверстия пищеварительной трубки. Изначально она заканчивается с обоих концов, однако к концу 8 недели происходит образование каналов, а слизистый эпителий закрывает просвет кишечной трубки.
Среди наиболее часто встречающихся патологий можно выделить стенозы (сужения или растяжки стенок) или атрезии (сращивания).
Больше всего страдает 12-перстная кишка, что связано с особенностями её эмбриогенеза. 1/2 случаев сопровождается пороками других внутренних органов — сердца, сосудов, прямой кишки, печени, желудка. Некоторые случаи настолько тяжёлые, что малышу при жизни придётся сделать множество операций, и они не будут являться гарантом его нормального существования.
Аномалии органов ЖКТ на УЗИ видны на сроке 11 недель. Ультразвуковая диагностика не является 100% гарантией того, что у малыша будут серьёзные отклонения, поэтому её результаты являются основанием для более детального обследования женщины.
Беременной делают кариотипирование на выявление хромосомных нарушений. Также она проходит анализ амниотической жидкости, и по результатам обследования (если они плохие и диагноз подтвердится) ей рекомендуют прервать беременность
Патологии кишечника
К аномалиям 12-перстной кишки относятся:
Атрезия. Встречается в 1 случае из 10 000. Заключается в полной непроходимости кишечника вследствие патологического сращения стенок органа. В 37% случаев сопровождается другими аномалиями — конской стопой, слиянием шейных позвонков, несимметричным положением рёбер и пр.
Ещё в 2% случаев атрезия кишечника сопровождается атрезией пищевода, гигромой заднего прохода, незавершённом поворотом желудка и т.п. В основном это типично для плода с хромосомными нарушениями, в частности с трисомией по 21 хромосоме.
90% беременностей заканчиваются выкидышем или замиранием развития в течение первых 2-х триместров. Остальные 10% беременностей с патологией 12-перстной кишки завершаются рождением детей, страдающих различными пороками: у 31% имеется обструкция дыхательных путей (закупорка инородным телом вроде кисты, опухоли), 24% — парез лицевого нерва (нарушением функциональности мимических мышц).
Только 1% малышей ведёт относительно нормальный образ жизни после проведения сложнейшей операции при условии отсутствия хромосомных нарушений.
Внутрикишечная мембрана . Это плёнка, перекрывающая просвет кишечника, появившаяся в результате нарушения разрастания внутреннего слоя 12-перстной кишки. Встречается в 1 случае из 40 000. На УЗИ визуализируется как слабоэхогенное образование. Просвет кишечника при этом сужен на несколько миллиметров, контуры слизистой оболочки чёткие.
Патология не является показанием для прерывания беременности. В зависимости от расположения мембраны она удаляется после рождения малыша методом дуоденотомии (вскрытием просвета кишечника с последующим удалением мембраны).
Мальротация. Заключается в нарушении нормального вращения и фиксации 12-перстной кишки. Если средняя кишка совершила полный оборот на кровоснобжающей ножке, это может привести к прекращению кровоснабжения и отмиранию средней кишки.
Пренатальный диагноз можно поставить с 24 недели, причём в 61,5% беременностей наблюдалось многоводие. На УЗИ выявляется анэхогенный double-buble 3 следствие расширение кишки и желудка.
Хотя даже незначительное расширение на сроке 16-22 недели должно вызывать тревогу. В норме 12-перстная кишка видна на УЗИ только с 24 недели. Дополнительно в 62% случаев выявляются у плода пороки развития сердца, мочеполовой системы, других органов ЖКТ. После исследования на кариотип в 67% случаев выявляются хромосомные отклонения, из которых на 1 месте стоит синдром Дауна.
Стеноз. Выявляется у 30% новорождённых, в основном у мальчиков. Это частичная непроходимость 12-перстного кишечника, локализованная в одном месте. В основном наблюдается в верхних отделах и сопровождается аномалиями поджелудочной железы. На УЗИ отчётливо виден на сроке от 24 недель при использовании допплеровского метода в изучении кровотока кишечника.
Стеноз успешно устраняется и имеет более благоприятные перспективы, чем атрезия. Не требует прерывания беременности.
Megaduodenum . Это увеличение размеров 12-перстной кишки до размеров, иногда превышающих размеры желудка. Встречается в 1 случае из 7500. Может являться следствием кольцевидной поджелудочной железы, когда головка органа кольцом окручивает кишечник, либо атрезии или стеноза 12-перстной кишки. На УЗИ диагностируется на 24 неделе. Верхняя часть брюшной полости вздута очень сильно, в то время как нижняя часть впалая.
Гиперэхогенность кишечника. Чем выше плотность исследуемой ткани, тем больше будет эхогенность. На УЗИ эхогенность кишечника плода должна быть ниже эхогенности костей, но выше, чем у таких пористых органов, как печень, лёгкие или почки. Когда эхогенность кишечника равна по плотности эхогенности костной ткани, говорят о гиперэхогенности.
Патология выявляется не ранее чем на 16 неделе. Она свидетельствует об отклонении в развитии плода. Повышенная эхогенность случается при преждевременном старении плаценты, внутренних инфекциях, несоответствии размеров плода сроку беременности, эндокринном заболевании муковисцидозе, кишечной непроходимости (стенозе).
УЗИ следует пройти в нескольких разных клиниках во избежание ошибки специалиста. Только при окончательном подтверждении диагноза женщину отправляют на более детальное обследование — биохимический скрининг, анализ на ТОРЧ-инфекции, кордоцентез и анализ амниотической жидкости. Окончательный диагноз ставится на основе комплексного анализа, а не только УЗИ обследования.
Дивертикулы (кисты). Они имеют разные названия — дупликационные кисты, удвоенная кишка, энтерогенная дивертикула. Заключается в отпочковании от стенок кишки образования в эмбриональный период. Образуются не только в кишечнике, но и по всему ЖКТ от гортани до ануса.
Считается, что причиной раздвоения стенок служит нарушение кровоснабжения пищеварительной трубки плода. Кисты на УЗИ гипоэхогенны, бывают как однокамерными, так и многокамерными. Стенки кист двухслойны и имеют повышенную перистальтику, имеют гиперэхогенность, если содержат кровь.
Визуализируются кисты кишечника на 2 триместре и часто сочетаются с другими патологиями. Точность визуализации кист кишечника у плода составляет 66,6%. Данная патология не является показанием к прерыванию беременности, потому что в неосложнённых случаях оперируется и устраняется.
Аномалия формы, размера, положения и подвижности кишечника. Ко 2 триместру беременности должен обратиться вокруг брыжеечной артерии против часовой стрелки на 2700. При нарушении эмбриогенеза можно выделить следующие патологии: отсутствие поворота, несостоявшийся поворот и неполный поворот.
На УЗИ при аномалиях поворота кишечника у плода отмечается многоводие и расширение петель кишечника без перистальтики. В случае перфорации кишечника возникает микониевый перитонит — заражение вследствие выхода наружу содержимого кишечника. Обнаруживается патология поздно, только на 3 триместре, что требует немедленной подготовки женщины к родоразрешению.
Патологии печени у плода
Печень визуализируется на ультразвуковом обследовании уже на 1-м скрининге. На сроке 11-14 недель можно увидеть в верхней части брюшной полости гипоэхогенное образование в виде месяца. К 25 неделе эхогенность повышается и становится такой же, как у кишечника, а перед родами превышает по плотности кишечник.
Очень важна оценка состояния кровотока печени. Вена пуповины плода входит в печень, во 2 триместре визуализируется воротная вена. Её диаметр в норме равен 2-3 мм, а к родам увеличивается до 10-11 мм. Желчные протоки в норме визуализироваться у плода не должны.
Одна из часто встречающихся патологий плода — гепатомегалия печени — увеличение размеров органа. Для выявления аномалии применяется 3D датчик, способный визуализировать срез в продольном, поперечном и вертикальном срезах. Также можно увидеть увеличение размеров печени и на обычном УЗИ аппарате по выступающему животику, охват которого значительно превышает норму.
Одновременно с этим на экране видны различные гиперэхогенные включения. Как правило, аномалия дополняется увеличением селезёнки.
Среди причин, приводящих к увеличению размеров органов пищеварения, выделяют скрытые инфекции (токсоплазмоз, сифилис, ветрянка), а также хромосомные мутации (синдромы Дауна, Зельвегера, Беквета-Видемана).
- При синдроме Зельвегера видны аномалии конечностей, искажение грудной клетки, кисты почек. Анализ амниотических вод выявляет нехватку дигидрокси-ацетон-фосфат-ацил-трансферазы.
- Ветряная оспа, герпес, цитомегаловирус вызывают кальцинирование тромбов печёночной вены, что отражается на экране УЗИ монитора гиперэхогенными кальцинатами круглой формы. Также они образуются при мекониевом перитоните — отравлении содержимым кишечника плода, которое попадает в результате повреждения стенок.
В 87,5% случаев причиной увеличения печени и образования кальцификатов являются внутриутробные инфекции. Также у большинства беременных диагностируется гиперэхогенный кишечник, изменение структуры плаценты, а также большие размеры селезёнки. Патология на 3 триместре возникает в случае резус-конфликта между матерью и ребёнком.
Также не исключены и метаболические нарушения. Увеличение печени встречается при галактоземии (генетическое нарушением углеводного обмена, из-за которого галактоза не преобразуется в глюкозу), трипсинемии (отсутствия выработки пищеварительного гормона трипсина), метилмалоновой ацидемии (отсутствии превращения D-метилмалоновой кислоты в янтарную кислоту), нарушениях выделения мочевины.
Одиночные гиперэхогенные включения большого размера гораздо лучше, чем множественные разрозненные образования в сочетании с другими патологиями. Практически в 100% случаев гиперэхогенные включения большого размера устраняются до рождения малыша или в первый год жизни.
В некоторых случаях порок органов брюшной полости ставится ошибочно. Такое бывает, если брюшная полость малыша сдавливается стенками матки, патологиях миометрия или других факторах.
На экране монитора видна псевдоомфалоцеле — ошибочная визуализация выхода органов брюшной полости за пределы брюшной стенки. Иногда УЗИ «не видит» значительных пороков. Так, грыжа по форме и эхоструктуре напоминает петли кишечника, в этом плане большую помощь оказывает допплерометрия, позволяющая увидеть кровоток.
Наша клиника имеет прекрасный 4D аппарат, оснащённый всеми современными возможностями, исключающими ошибки диагностики.
Желудок
На 16-20 неделе визуализируется желудок плода как анэхогенное образование круглой или овальной формы в верхних отделах брюшной полости. Если желудок не наполнен амниотическими водами, то можно говорить об атрезии пищевода (полном отсутствии просвета).
При диафрагмальной грыже желудок смещён и также не может быть определён на УЗИ. Также амниотическая жидкость отсутствует при поражении ЦНС у плода.
Если плод заглатывает вместе с жидкостью кровь, в желудке визуализируются гиперэхогенные включения. Они также видны при опухолях желудка, но они обычно сопровождаются другими пороками развития. Размеры органа увеличиваются при кишечной непроходимости, многоводии, утолщении стенок, отсутствии малой кривизны.
Уменьшение размеров желудка типично для микрогастрии, которая возникает на фоне отсутствия мочевого пузыря или неправильного положения печени. В 52% случаев плод погибает до 24-й недели беременности, ребёнок рождается нежизнеспособным.
Щелевидный желудок характерен для недоразвитии на ранних стадиях беременности. Данная патология исправляется после рождения малыша: ребёнку конструируют желудок из части тонкого кишечника. Операция крайне сложная, но аномалия не является показанием для выполнения аборта.
Атрезия желудка характеризуется отсутствием эхотени и предполагает образование плёнки с отверстием или без него, расположенной поперёк стенок желудка. Если это изолированная патология, то в 90% случаев она устраняется хирургически. Но обычно атрезия желудка сочетается со сращиванием пищевода, асцитом (излишним скоплением жидкости), недоразвитостью лёгких.
Агенезия желудка предполагает полное отсутствие органа. Это типично для тяжёлых хромосомных аномалий, от которых плод погибает в пренатальный период. Большое значение в диагностике имеет УЗИ на 22 неделе. некоторые отклонения исчезают сами собой, а некоторые требуют немедленного вмешательства.
Лицевые дефекты - протезирование при синдроме Гольденхара

Синдром Гольденхара (гемифациальной микросомии) - редко встречающаяся врожденная патология, которая проявляется неправильным развитием одной половины лица и позвоночника. При синдроме Гольденхара наблюдается классическая триада симптомов - гипоплазия нижней челюсти, неправильное формирование глаза и ушной раковины, а также отклонение в строении шейных позвонков.
Проявления синдрома Гольденхара
У детей с синдромом Гольденхара наблюдается серьезный дефект лица - одна половина лица меньше, недоразвиты верхняя и нижняя челюсти и мимическая мускулатура. Недоразвитая, низкорасположенная ушная раковина, узкий наружный слуховой проход, дефекты барабанной перепонки, проводниковая тугоухость или глухота. Ухо иногда может отсутствовать. Отмечается рот больше средних размеров, один угол рта выше другого. Парез лицевого нерва. Глаз недоразвит или полностью отсутствует. Аномальное строение шейных позвонков, их сращение, шея короткая.
Часто наблюдаются при синдроме Гольденхара аномалии развития сердечно-сосудистой, мочеполовой и центральной нервной системы.
Диагностика
Диагностика начинается с составления анамнеза и тщательного опроса родителей больного ребенка. Часто наследственный характер заболевания подтверждается наличием данного синдрома у кого-нибудь из родственников. Помимо аномалий лица, у пациентов нарушена работа многих внутренних органов. Для диагностики степени их поражения назначается полное обследование при помощи инструментальных методов - МРТ, КТ, УЗИ, а также проводится генетическое исследование.
Лечение синдрома Гольденхара
Так как проявления синдрома Гольденхара могут быть самыми различными, в лечении такжевозможны варианты от простого наблюдения до многоэтапных сложных операций.
Поскольку выявляется синдром Гольденхара при рождении ребенка, то и лечение можно начинать сразу же, как только малыш окрепнет. Для реконструкции лица и черепа, при необходимости, проводят пластическую операцию. Дети с синдромом Гольденхара имеют множество других врожденных патологий, для их лечения привлекаются врачи различных специальностей, на консилиуме разрабатывается индивидуальный план лечения ребенка.
Если своевременное лечение не было получено, с возрастом асимметрия лица будет прогрессировать (дефект лиц, все больше обезображивая внешность и усугубляя психологическую травму.
В таких случаях коррекция при помощи лицевого протезирования поможет скрыть лицевые дефекты и адаптировать пациента к жизни в обществе.
Читайте также: