УЗИ, МРТ при синдроме PHACES у плода
Добавил пользователь Alex Обновлено: 01.02.2026
Лысова Ю.В.
Научный руководитель: к.м.н., доцент Нечаев В.Н.
Резюме
Количество врожденных пороков развития в последнее десятилетие заметно увеличилось. Пороки развития нервной системы суммарно занимают третье место в структуре аномалий развития после врожденной патологии сердечно-сосудистой и мочевыводящей систем, причем около 80% этих заболеваний представлены гидроцефалией различного генеза [1]. Среди большого количества возможных аномалий одним из наиболее тяжелых по своим последствиям считается синдром Денди Уокера.
Ключевые слова
Статья
Данный синдром был впервые описан американским нейрохирургом Уолтером Денди в 1921 году и Эрлом Уокером в 1944 г. Среди живорождённых детей частота встречаемости от 1:5000 до 1:25000, а среди детей с врождённой гидроцефалией колеблется от 3,5 до 12% [2].
Синдром Денди-Уокера (Dandy-Walker Syndrome) - это порок развития головного мозга (мозжечка и окружающих его ликворных пространств), для которого характерна триада симптомов: гипотрофия червя мозжечка и/или полушарий мозжечка, кисты задней черепной ямки, гидроцефалия различной степени. Данный синдром по МКБ Х кодируется в рубрике аномалий развития под шифром Q 03.1, как атрезия отверстий Мажанди и Люшка [3, 4].
Согласно современным представлениям этиология синдрома Денди-Уокера чрезвычайно гетерогенна, так как в его возникновении принимают участие разные факторы: наследственные (хромосомные и генные) и экзогенные тератогены; предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха); приём алкоголя, диабет беременно. В 1/3 - 1/2 случаев синдром Денди-Уокера сочетается с другими различными врожденными синдромами (табл. 1) [2].
В 70% случаев порок сочетается и с другими аномалиями головного мозга — агенезией мозолистого тела, энцефалоцеле, полимикрогирией, агирией, гетеротопией серого вещества, а также с поражениями других органов и систем (полидактилией, синдактилией, врожденными пороками сердца, поликистозом почек, расщелинами неба и др.) [5].
Среди основных гипотез возникновения синдрома Денди-Уокера можно выделить следующие:
- остановка эмбрионального развития в процессе формирования ромбовидного мозга;
- атрезия выходного отверстия из IV желудочка при отсроченном открытии отверстия Мажанди;
- возникновение сосудистого сплетения IV желудочка в середине тонкой крыши ромбовидного мозга.
Синдром Денди-Уокера характеризуется выраженным клиническим полиморфизмом: от практически нормального постнатального развития до тяжелой инвалидности и даже гибели ребенка. Перинатальные исходы во многом зависят от глубины поражения ЦНС (прогрессирующая гидроцефалия), а также от наличия сочетанной патологии, которая в 60 - 75% случаев сопровождает данный синдром [2, 5].
Прогноз для жизни и здоровья при синдроме Денди — Уокера зависит от наличия сочетанных аномалий развития, хромосомных аномалий и срока диагностики. По данным литературы, показатели постнатальной заболеваемости и смертности выше в тех случаях, когда синдром диагностирован в пренатальном периоде, а не постнатально [2] .
Лечение данного заболевания симптоматическое. При наличии признаков нарастающей внутричерепной гипертензии проводят шунтирующие операции. Исход часто летальный, в 90% случаев это первые годы жизни [7].
Под наблюдением находился доношенный новорождённый мальчик Л., родившийся от 3 беременности, протекавшей на фоне поздней постановки на учёт (в 30 недель). С 35 недель гестации отмечалось многоводие и впервые по данным ультразвукового исследования выявлена аномалия развития плода - мальформация Денди-Уокера.
Картина пренатального УЗИ в 35 недель гестации: форма головы аномальная (клубникообразная). Кости при надавливании датчиком не деформируются. Расширение боковых желудочков, передние рога до 10 мм справа и слева. Полость прозрачной перегородки до 9,9 мм. Задние рога - 11 мм, слева - 12 мм. Отмечено расширение IVжелудочка до 12 мм. Имеется гипоплазия червя мозжечка, расширение большой цистерны до 12 мм.
Ребенок от третьих срочных родов в головном предлежании. Околоплодные воды светло-жёлтые в объеме двух литров. Масса тела ребёнка при рождении составила 2890 г, рост - 49 см., окружность головы - 35 см., груди - 33 см. Оценка по шкале Апгар 4 - 7 баллов.
Состояние ребёнка после рождения тяжелое, из родильного зала поступил в ОРИТН, за счёт дыхательной недостаточности III степени и выраженной неврологической симптоматики. C момента поступления находился на ИВЛ, с третьих суток жизни был переведен на респираторную поддержку в режиме СРАР, а с 7 суток жизни на спонтанное дыхание. Оксигенотерапия проводилась по показаниям. Энтеральное питание начато с первых суток в трофическом объеме с последующим расширением, усваивал.
Неврологически: положение ребёнка вынужденное на боку с запрокинутой головой, крик монотонный, имеются плавающие движения глазных яблок, симптом «заходящего солнца», непостоянный горизонтальный нистагм. Неправильная форма черепа, увеличенная в размерах мозговая часть, нарастание окружности головы в динамике за 2 недели составило 2,5 см. Отмечалось расхождение костей черепа, швы открыты до 0,5 см, с увеличением размеров большого родничка до 5 × 8 см. Зрачки средней величины, фотореакция сохранена. Спонтанная двигательная активность и мышечный тонус снижены. Дистония в конечностях. Физиологические рефлексы угнетены, сухожильные снижены, симметричные.
Обращало на себя внимание наличие двусторонней косолапости с варусной деформацией обеих стоп, кистей и фаланг пальцев, гипертелоризма, седловидной переносицы, нависающего лба и затылка, широкого языка, короткой шеи (рис. 3).
На 14 сутки жизни, после стабилизации состояния, ребёнок был переведён на второй этап лечения, в отделение патологии новорожденных для дальнейшего наблюдения и лечения.
Картина нейросонографии в первые сутки жизни: в задней черепной ямке при коронарных и сагиттальном сканированиях наблюдается крупное анэхогенное («кистовидное») образование, включающее расширенные III и IV желудочек; полушария мозжечка резко уменьшены, червь не определяется; мозжечковый намет смещен вверх.
В динамике по данным НСГ выявлена перивентрикулярная ишемия, подтверждён синдром Денди-Уокера, вентрикуломегалия (как часть симптомокомплекса) и повышенная резистентность сосудов мозга.
Ребёнок был осмотрен неврологом, окулистом, генетиком, выставлен диагноз: Врожденный порок развития ЦНС - синдром Денди Уокера. Гипоксически-ишемическое поражение ЦНС. После осмотра нейрохирурга выставлен диагноз: ВПР ЦНС синдром Денди-Уокера, гидроцефальный синдром.
На момент осмотра в нейрохирургическом лечении не нуждался, были даны рекомендации по уходу и лечению.
По результатам ДЭХО-КГ выявлен врожденный порок сердца: комбинированный стеноз легочной артерии (клапанно-подклапанный), ДМПП со сбросом слева направо. Открытое овальное окно диаметром 2,0 мм.
Ребенок был консультирован кардиологом и кардиохирургом, даны рекомендации.
По данным УЗИ брюшной полости и почек патологии не выявлено.
После осмотра врача-ортопеда была подтверждена врожденная двусторонняя косолапость.
В связи со стабилизацией состояния ребёнка, после проведённого обследования и лечения, а также отказа матери от родительских прав, мальчик на 57 сутки жизни был переведён в дом ребёнка города Маркса.
В заключение следует сказать о том, что проведённое настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку встречается ни так часто в повседневной практике врача.
Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое клиническое наблюдение, представляет определенные сложности. По моему мнению, в подобных ситуациях оправдана постановка синдромального диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования. Точный нозологический диагноз важен не только для генетического анализа, медико-генетического консультирования, но и, прежде всего, для профилактики и лечения. Без достоверного клинического диагноза невозможен анализ факторов и их теоретическое осмысление.
Литература
2. Барашнев Ю.И. Перинатальная неврология. - М.: Триада-Х, 2001.- С. 181-232.
4. Неонатология (национальное руководство) / Под ред. Н.И.Володина. - М., ГЭОТАР-Медиа, 2007; 847 с.
5. Наследственные болезни / Под ред. Л.О.Бадаляна. - Т.: Медицина, 2002.- С. 138.
6. Пренатальная диагностика наследственных и врожденных болезней / Под ред. Э.К.Айламазяна, В.С.Баранова. - М.: Триада-Х, 2007. - С. 11-148.
7. Барашнев Ю.И., Бахарев В.А., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний у детей. М.: Триада-Х, 2004.- С.12-87.
Таблицы
Синдромы, с которыми сочетается гидроцефалия Денди-Уокера
Синдромы
Клинические признаки
Этиология
Различные хромосомные аномалии
Поражение различных органов и систем
Хромосомные делеции, дупликации, трисомии, триплоидии
Лиссэнцефалия, ретинальная дисплазия, аномалия глаз, энцефалоцеле, миопатии и др.
Мутации генов POMT1 (локализации 9g34.1), POMT2 (14g24.3), FKTN (9g31) и др. Тип наследования аутосомно-рецессивный
3С синдром (краниомозжечково-сердечная дисплазия, Ritscher-Schinzel синдром)
Задержка роста, пороки сердца (септальные дефекты), гипертелоризм и др.
Ген не установлен. Тип наследования аутосомно-рецессивный
Артериальные аномалии, включая коарктацию аорты, дефекты сердца, глаз (микроофтальмия).
Синдром Денди-Уокера
Синдром Денди-Уокера - это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.
МКБ-10



Общие сведения
Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.

Причины
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Патогенез
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Классификация
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
В 1989 г. американским педиатром и нейрорадиологом Джеймсом Барковичем была предложена альтернативная классификация, базирующаяся на результатах МР-диагностики. Выделена группа заболеваний, названная «комплексом Денди-Уокера», который включает 4 клинических формы: классическую аномалию (полная форма), вариант Денди-Уокера (менее грубые нарушения), кисту кармана Блейка и mega cisterna magna.
Симптомы
Основное проявление синдрома Денди-Уокера - гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера - симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
Осложнения
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Диагностика
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
Дифференциальная диагностика
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
Хирургическое лечение
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Прогноз и профилактика
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. - 2018. - №2.
2. Аномалия Денди-Уокера - редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. - 2017. - №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. - 2010. - №1.
Аномалии и патологии органов пищеварительной системы плода, выявляемые на УЗИ
Патологии органов пищеварительной системы встречаются у плода нередко как самостоятельно, так и в комплексе с другими аномалиями внутренних органов. На них приходится до 21% пороков у новорождённых и 34% случаев младенческой смертности.
Причины нарушений формирования органов ЖКТ у плода, статистика
Аномалии строения органов ЖКТ связаны с нарушением эмбриогенеза на стадии 4-8 недель беременности, когда идёт образование отверстия пищеварительной трубки. Изначально она заканчивается с обоих концов, однако к концу 8 недели происходит образование каналов, а слизистый эпителий закрывает просвет кишечной трубки.
Среди наиболее часто встречающихся патологий можно выделить стенозы (сужения или растяжки стенок) или атрезии (сращивания).
Больше всего страдает 12-перстная кишка, что связано с особенностями её эмбриогенеза. 1/2 случаев сопровождается пороками других внутренних органов — сердца, сосудов, прямой кишки, печени, желудка. Некоторые случаи настолько тяжёлые, что малышу при жизни придётся сделать множество операций, и они не будут являться гарантом его нормального существования.
Аномалии органов ЖКТ на УЗИ видны на сроке 11 недель. Ультразвуковая диагностика не является 100% гарантией того, что у малыша будут серьёзные отклонения, поэтому её результаты являются основанием для более детального обследования женщины.
Беременной делают кариотипирование на выявление хромосомных нарушений. Также она проходит анализ амниотической жидкости, и по результатам обследования (если они плохие и диагноз подтвердится) ей рекомендуют прервать беременность
Патологии кишечника
К аномалиям 12-перстной кишки относятся:
Атрезия. Встречается в 1 случае из 10 000. Заключается в полной непроходимости кишечника вследствие патологического сращения стенок органа. В 37% случаев сопровождается другими аномалиями — конской стопой, слиянием шейных позвонков, несимметричным положением рёбер и пр.
Ещё в 2% случаев атрезия кишечника сопровождается атрезией пищевода, гигромой заднего прохода, незавершённом поворотом желудка и т.п. В основном это типично для плода с хромосомными нарушениями, в частности с трисомией по 21 хромосоме.
90% беременностей заканчиваются выкидышем или замиранием развития в течение первых 2-х триместров. Остальные 10% беременностей с патологией 12-перстной кишки завершаются рождением детей, страдающих различными пороками: у 31% имеется обструкция дыхательных путей (закупорка инородным телом вроде кисты, опухоли), 24% — парез лицевого нерва (нарушением функциональности мимических мышц).
Только 1% малышей ведёт относительно нормальный образ жизни после проведения сложнейшей операции при условии отсутствия хромосомных нарушений.
Внутрикишечная мембрана . Это плёнка, перекрывающая просвет кишечника, появившаяся в результате нарушения разрастания внутреннего слоя 12-перстной кишки. Встречается в 1 случае из 40 000. На УЗИ визуализируется как слабоэхогенное образование. Просвет кишечника при этом сужен на несколько миллиметров, контуры слизистой оболочки чёткие.
Патология не является показанием для прерывания беременности. В зависимости от расположения мембраны она удаляется после рождения малыша методом дуоденотомии (вскрытием просвета кишечника с последующим удалением мембраны).
Мальротация. Заключается в нарушении нормального вращения и фиксации 12-перстной кишки. Если средняя кишка совершила полный оборот на кровоснобжающей ножке, это может привести к прекращению кровоснабжения и отмиранию средней кишки.
Пренатальный диагноз можно поставить с 24 недели, причём в 61,5% беременностей наблюдалось многоводие. На УЗИ выявляется анэхогенный double-buble 3 следствие расширение кишки и желудка.
Хотя даже незначительное расширение на сроке 16-22 недели должно вызывать тревогу. В норме 12-перстная кишка видна на УЗИ только с 24 недели. Дополнительно в 62% случаев выявляются у плода пороки развития сердца, мочеполовой системы, других органов ЖКТ. После исследования на кариотип в 67% случаев выявляются хромосомные отклонения, из которых на 1 месте стоит синдром Дауна.
Стеноз. Выявляется у 30% новорождённых, в основном у мальчиков. Это частичная непроходимость 12-перстного кишечника, локализованная в одном месте. В основном наблюдается в верхних отделах и сопровождается аномалиями поджелудочной железы. На УЗИ отчётливо виден на сроке от 24 недель при использовании допплеровского метода в изучении кровотока кишечника.
Стеноз успешно устраняется и имеет более благоприятные перспективы, чем атрезия. Не требует прерывания беременности.
Megaduodenum . Это увеличение размеров 12-перстной кишки до размеров, иногда превышающих размеры желудка. Встречается в 1 случае из 7500. Может являться следствием кольцевидной поджелудочной железы, когда головка органа кольцом окручивает кишечник, либо атрезии или стеноза 12-перстной кишки. На УЗИ диагностируется на 24 неделе. Верхняя часть брюшной полости вздута очень сильно, в то время как нижняя часть впалая.
Гиперэхогенность кишечника. Чем выше плотность исследуемой ткани, тем больше будет эхогенность. На УЗИ эхогенность кишечника плода должна быть ниже эхогенности костей, но выше, чем у таких пористых органов, как печень, лёгкие или почки. Когда эхогенность кишечника равна по плотности эхогенности костной ткани, говорят о гиперэхогенности.
Патология выявляется не ранее чем на 16 неделе. Она свидетельствует об отклонении в развитии плода. Повышенная эхогенность случается при преждевременном старении плаценты, внутренних инфекциях, несоответствии размеров плода сроку беременности, эндокринном заболевании муковисцидозе, кишечной непроходимости (стенозе).
УЗИ следует пройти в нескольких разных клиниках во избежание ошибки специалиста. Только при окончательном подтверждении диагноза женщину отправляют на более детальное обследование — биохимический скрининг, анализ на ТОРЧ-инфекции, кордоцентез и анализ амниотической жидкости. Окончательный диагноз ставится на основе комплексного анализа, а не только УЗИ обследования.
Дивертикулы (кисты). Они имеют разные названия — дупликационные кисты, удвоенная кишка, энтерогенная дивертикула. Заключается в отпочковании от стенок кишки образования в эмбриональный период. Образуются не только в кишечнике, но и по всему ЖКТ от гортани до ануса.
Считается, что причиной раздвоения стенок служит нарушение кровоснабжения пищеварительной трубки плода. Кисты на УЗИ гипоэхогенны, бывают как однокамерными, так и многокамерными. Стенки кист двухслойны и имеют повышенную перистальтику, имеют гиперэхогенность, если содержат кровь.
Визуализируются кисты кишечника на 2 триместре и часто сочетаются с другими патологиями. Точность визуализации кист кишечника у плода составляет 66,6%. Данная патология не является показанием к прерыванию беременности, потому что в неосложнённых случаях оперируется и устраняется.
Аномалия формы, размера, положения и подвижности кишечника. Ко 2 триместру беременности должен обратиться вокруг брыжеечной артерии против часовой стрелки на 2700. При нарушении эмбриогенеза можно выделить следующие патологии: отсутствие поворота, несостоявшийся поворот и неполный поворот.
На УЗИ при аномалиях поворота кишечника у плода отмечается многоводие и расширение петель кишечника без перистальтики. В случае перфорации кишечника возникает микониевый перитонит — заражение вследствие выхода наружу содержимого кишечника. Обнаруживается патология поздно, только на 3 триместре, что требует немедленной подготовки женщины к родоразрешению.
Патологии печени у плода
Печень визуализируется на ультразвуковом обследовании уже на 1-м скрининге. На сроке 11-14 недель можно увидеть в верхней части брюшной полости гипоэхогенное образование в виде месяца. К 25 неделе эхогенность повышается и становится такой же, как у кишечника, а перед родами превышает по плотности кишечник.
Очень важна оценка состояния кровотока печени. Вена пуповины плода входит в печень, во 2 триместре визуализируется воротная вена. Её диаметр в норме равен 2-3 мм, а к родам увеличивается до 10-11 мм. Желчные протоки в норме визуализироваться у плода не должны.
Одна из часто встречающихся патологий плода — гепатомегалия печени — увеличение размеров органа. Для выявления аномалии применяется 3D датчик, способный визуализировать срез в продольном, поперечном и вертикальном срезах. Также можно увидеть увеличение размеров печени и на обычном УЗИ аппарате по выступающему животику, охват которого значительно превышает норму.
Одновременно с этим на экране видны различные гиперэхогенные включения. Как правило, аномалия дополняется увеличением селезёнки.
Среди причин, приводящих к увеличению размеров органов пищеварения, выделяют скрытые инфекции (токсоплазмоз, сифилис, ветрянка), а также хромосомные мутации (синдромы Дауна, Зельвегера, Беквета-Видемана).
- При синдроме Зельвегера видны аномалии конечностей, искажение грудной клетки, кисты почек. Анализ амниотических вод выявляет нехватку дигидрокси-ацетон-фосфат-ацил-трансферазы.
- Ветряная оспа, герпес, цитомегаловирус вызывают кальцинирование тромбов печёночной вены, что отражается на экране УЗИ монитора гиперэхогенными кальцинатами круглой формы. Также они образуются при мекониевом перитоните — отравлении содержимым кишечника плода, которое попадает в результате повреждения стенок.
В 87,5% случаев причиной увеличения печени и образования кальцификатов являются внутриутробные инфекции. Также у большинства беременных диагностируется гиперэхогенный кишечник, изменение структуры плаценты, а также большие размеры селезёнки. Патология на 3 триместре возникает в случае резус-конфликта между матерью и ребёнком.
Также не исключены и метаболические нарушения. Увеличение печени встречается при галактоземии (генетическое нарушением углеводного обмена, из-за которого галактоза не преобразуется в глюкозу), трипсинемии (отсутствия выработки пищеварительного гормона трипсина), метилмалоновой ацидемии (отсутствии превращения D-метилмалоновой кислоты в янтарную кислоту), нарушениях выделения мочевины.
Одиночные гиперэхогенные включения большого размера гораздо лучше, чем множественные разрозненные образования в сочетании с другими патологиями. Практически в 100% случаев гиперэхогенные включения большого размера устраняются до рождения малыша или в первый год жизни.
В некоторых случаях порок органов брюшной полости ставится ошибочно. Такое бывает, если брюшная полость малыша сдавливается стенками матки, патологиях миометрия или других факторах.
На экране монитора видна псевдоомфалоцеле — ошибочная визуализация выхода органов брюшной полости за пределы брюшной стенки. Иногда УЗИ «не видит» значительных пороков. Так, грыжа по форме и эхоструктуре напоминает петли кишечника, в этом плане большую помощь оказывает допплерометрия, позволяющая увидеть кровоток.
Наша клиника имеет прекрасный 4D аппарат, оснащённый всеми современными возможностями, исключающими ошибки диагностики.
Желудок
На 16-20 неделе визуализируется желудок плода как анэхогенное образование круглой или овальной формы в верхних отделах брюшной полости. Если желудок не наполнен амниотическими водами, то можно говорить об атрезии пищевода (полном отсутствии просвета).
При диафрагмальной грыже желудок смещён и также не может быть определён на УЗИ. Также амниотическая жидкость отсутствует при поражении ЦНС у плода.
Если плод заглатывает вместе с жидкостью кровь, в желудке визуализируются гиперэхогенные включения. Они также видны при опухолях желудка, но они обычно сопровождаются другими пороками развития. Размеры органа увеличиваются при кишечной непроходимости, многоводии, утолщении стенок, отсутствии малой кривизны.
Уменьшение размеров желудка типично для микрогастрии, которая возникает на фоне отсутствия мочевого пузыря или неправильного положения печени. В 52% случаев плод погибает до 24-й недели беременности, ребёнок рождается нежизнеспособным.
Щелевидный желудок характерен для недоразвитии на ранних стадиях беременности. Данная патология исправляется после рождения малыша: ребёнку конструируют желудок из части тонкого кишечника. Операция крайне сложная, но аномалия не является показанием для выполнения аборта.
Атрезия желудка характеризуется отсутствием эхотени и предполагает образование плёнки с отверстием или без него, расположенной поперёк стенок желудка. Если это изолированная патология, то в 90% случаев она устраняется хирургически. Но обычно атрезия желудка сочетается со сращиванием пищевода, асцитом (излишним скоплением жидкости), недоразвитостью лёгких.
Агенезия желудка предполагает полное отсутствие органа. Это типично для тяжёлых хромосомных аномалий, от которых плод погибает в пренатальный период. Большое значение в диагностике имеет УЗИ на 22 неделе. некоторые отклонения исчезают сами собой, а некоторые требуют немедленного вмешательства.
Синдром ЕЕС. Возможности пренатальной диагностики и особенности медико-генетического консультирования

Революционные изменения в экспертной диагностике. Безупречное качество изображения, молниеносная скорость работы, новое поколение технологий визуализации и количественного анализа данных УЗ-сканирования.
Введение
Синдром ЕЕС (ectrodactyly-ectodermal dysplasia-clefting syndrome) - редкий генетический синдром, обычно проявляющийся триадой признаков: расщелиной лица, эктродактилией конечностей и признаками эктодермальной дисплазии [1]. Выделяют 2 типа синдрома: ЕЕС-1 и ЕЕС-3. Ранее был описан также и ЕЕС-2, однако на сегодняшний день локализация гена, кодирующего ЕЕС-2, считается ошибочной, следовательно, и формы ЕЕС-2 не существует. Первый тип характеризуется мутацией в 7-й хромосоме в области 7q11.2-q21.3, третий - имеет в большинстве случаев мутацию Тp63-гена и другие новые, спонтанные мутации на 3-й хромосоме 3q27 [2]. В OMIM (ежедневно обновляемом международном классификаторе Online Mendelian Inheritance in Man), где представлены все известные на сегодняшний день фенотипы и генотипы менделирующих (наследуемых) болезней человека, эти виды синдрома кодируются по-разному: ЕЕС-1 - 129900, а ЕЕС-3 - 604292 [3].
ЕЕС-синдром - наследственная патология с аутосомно-доминантным типом наследования, характеризующаяся различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности) [1].
Манифестные признаки синдрома ЕЕС (синдромальное ядро)
Эктродактилия - это расщелина кистей и/или стоп с отсутствием одного или нескольких центральных пальцев, известная также под названием "Split hand-split foot malformation" (SHFM). Часто внешний вид кистей и стоп сравнивают с клешней краба. Нередко встречается в сочетании с синдактилией (сращением пальцев).
Эктодермальная дисплазия проявляется особенностями строения всех производных эктодермы. Изменения затрагивают волосы, зубы, ногти [11]. Волосы и ресницы у таких людей тусклые, редкие и жесткие. Характерны аномалии зубов, гипоплазия зубной эмали, снижение пигментации волос, кожи, радужной оболочки глаз, что вызывает светлый их цвет. Также часто сочетание синдрома с фотобоязнью [1], обструкцией носо-слезных каналов [12], кондуктив- ной тугоухостью [13].
Эктодермальная дисплазия проявляется отсутствием потовых желез, что характеризуется наличием гипогидротинового синдрома, проявляющегося в резкой сухости и шелушении кожи. Характерным проявлением этого синдрома является хриплый, осипший голос из-за нарушения увлажнения голосовых связок, вследствие чего не происходит полного их смыкания [14].
Нарушение речи у больных с ЕЕС-синдромом имеет много причин. Во-первых, это связано с последствиями наличия расщелины губы и/или неба и "гиперназальной" речью, во-вторых, правильной артикуляции может препятствовать не только изменение голосовых связок, но и патология зубов. В-третьих, из-за аномалий слуховых косточек часто прогрессирует тугоухость, ведущая к когнитивным расстройствам и расстройствам речи.
Дети с этим синдромом обычно имеют нормальный уровень интеллекта. Лечение основано на коррекции пороков лица и конечностей. Признаки эктодермальной дисплазии проявляются в постнатальном периоде [1]. Врожденные пороки развития (ВПР) мочевыделительной системы у больных с синдромом ЕЕС возникают более чем в половине случаев. Среди них описаны: мегауретер, гидронефроз, уретероцеле, аплазия почки, аномалии гениталий, уретровезикальный рефлюкс с частыми инфекциями мочевыделительных путей [12].
Пренатальная диагностика синдрома ЕЕС
Учитывая выраженность проявлений при синдроме ЕЕС, пренатальная диагностика этой патологии, безусловно, возможна. Однако на сегодняшний день в мировой литературе встречается незначительное число публикаций, посвященных диагностике этого редкого синдрома 16, что связано, видимо, с редкостью возникновения данной аномалии. В основном сроками постановки диагноза являются 16-30 недель беременности. Большую помощь в диагностике данной патологии, учитывая выраженные изменения фенотипа лица и конечностей, оказывает применение новых ультразвуковых технологий 3D/4D с методиками поверхностной реконструкции [16].
Первое описание пренатальной диагностики синдрома датировано 1993 годом и принадлежит M. Bronshtein и R. Gershoni, когда при применении трансвагинальной эхографии у плода была выявлена расщелина губы и неба и эктродактилия в 14 недель беременности [17]. Уникальность этого факта в том, что, пожалуй, впервые из известных случаев диагностики врожденных аномалий дебют дородового выявления порока принадлежит сроку первого триместра. Примечательно и то, что авторами не только описаны отдельно выявленные пороки развития плода, но и пренатально установлена нозология этого состояния. Другими словами, диагноз не звучал, как "множественные врожденные пороки развития", когда невозможно говорить об этиологии заболевания, а следовательно, и о мерах специфической профилактики данной патологии в дальнейшем в семье. Пренатальный диагноз был выставлен полно и абсолютно корректно, что особенно важно при проведении медико-генетического консультирования с формированием тактики адекватного репродуктивного поведения семьи в дальнейшем.
Из-за незначительного количества публикаций, посвященных пренатальной диагностике этого генетического синдрома, представляем ряд собственных наблюдений диагностики синдрома ЕЕС в разные сроки беременности, в том числе и в первом триместре. Особо рассмотрим особенности проведения медико-генетического консультирования при диагностике различных форм синдрома с аутосомно-доминантным типом наследования для выработки специфических мер профилактики данного наследственного заболевания.
Клиническое наблюдение 1
Пациентка Н. 24 лет. Беременность первая. Женщина и муж соматически здоровы, брак неродственный. Обратилась в медико-генетическое отделение МОНИИАГ в 21,4 недели беременности в связи с подозрением на порок развития конечностей у плода. Были выявлены: эктродактилия кистей (рис. 1) и стоп плода (рис. 2.). Из особенностей строения выявлены двусторонние пиелоэктазии (рис. 3). Проведено медико-генетическое консультирование. Семья приняла решение прервать данную беременность в связи с наличием инвалидизирующих пороков конечностей. При патологоанатомическом исследовании ВПР конечностей подтверждены, а также дополнительно выявлена расщелина мягкого неба и признаки эктодермальной дисплазии (гипертрофия десен, характерный лицевой фенотип). Окончательный диагноз: "синдром ЕЕС, аутосомно-доминантный тип наследования". Мутация de novo.
Оценка носовой кости в I триместре беременности: как, где, когда и зачем мы делаем
Московский областной НИИ акушерства и гинекологии, Москва.
Кафедра медицинской генетики РМАПО, Москва.
УЗИ аппарат HS40
Лидер продаж в высоком классе. Монитор 21,5" высокой четкости, расширенный кардио пакет (Strain+, Stress Echo), экспертные возможности для 3D УЗИ в акушерско-гинекологической практике (STIC, Crystal Vue, 5D Follicle), датчики высокой плотности.
Задача пренатального скрининга - выявление беременных женщин группы высокого риска по рождению детей с хромосомными болезнями и врожденными пороками развития с целью более детального анализа состояния плода с помощью специальных методов [1].
С конца прошлого века в алгоритм пренатального скрининга включен расчет индивидуального комбинированного риска, центральное место в котором занимают ультразвуковой и биохимический скрининг в I триместре (11-14 нед беременности). Были созданы компьютерные программы расчета риска, учитывающие возраст, ультразвуковой маркер I триместра (толщина воротникового пространства - ТВП) и биохимические маркеры крови (β-hCG и PAPP-A) беременной женщины [2].
За последние 10 лет данная система полностью оправдала себя и получила дальнейшее развитие, путем прибавления к расчету риска добавочных ультразвуковых маркеров (оценка носовой кости, венозного протока, трикуспидальной регургитации, некоторых маркерных врожденных пороков развития). Расширение протокола осмотра с оценкой новых ультразвуковых маркеров (оценка носовой кости, кровоток в венозном протоке и на трикуспидальном клапане) улучшает чувствительность комбинированного скрининга благодаря увеличению частоты обнаружения и уменьшению частоты ложноположительных результатов [3].
Однако их оценка требует соответствующего углубленного обучения врача УЗД и получение сертификата компетентности на проведение данного вида исследования, так как только после получения доступа на конкретный вид исследования программа расчета риска будет учитывать эти данные в своих расчетах 3.
Преимуществами проведения УЗИ в 11-14 нед помимо установки точного срока беременности являются: ранняя диагностика многих пороков развития плода, оценка маркеров хромосомных аномалий для выявления беременных высокого риска по хромосомным аномалиям у плода, при многоплодной беременности именно в ранний срок возможно установить хориальность, что является важнейшим фактором, определяющим исход многоплодной беременности, возможность выявить женщин группы высокого риска по развитию преэклампсии в поздние сроки беременности [3, 4].
Копчико-теменной размер плода (КТР) для проведения скрининга I триместра должен быть в пределах 45-84 мм. Для оценки носовой кости в I триместре беременности необходимо соблюдать строгие условия. Это адекватное увеличение (на снимке должны быть только голова и верхняя часть грудной клетки), среднесагиттальный скан (должны быть визуализированы эхогенный кончик носа, небный отросток верхней челюсти, диэнцефалон), нос представлен тремя "К" (кончик носа, кожа, кость). Кожные покровы и кости носа визуализируются в виде знака "равенства", нос параллелен датчику.
Такие правила, как размер плода, адекватное увеличение, среднесагиттальный скан идентичны таковым при измерении ТВП. Таким образом, при выведении корректного скана для измерения ТВП, что является обязательным при проведении УЗ-исследования в сроки 11-14 нед беременности, оценка носовой кости проводится в том же самом срезе, не требуя получения дополнительных изображений.
Если все критерии соблюдены, то на уровне носа плода должны быть видны три четко различимые линии: верхняя линия представляет собой кожу, книзу от нее визуализируется более толстая и более эхогенная, чем кожа носовая кость. Третья линия, визуализируемая кпереди от носовой кости и на более высоком уровне, чем кожа - это кончик носа (рис. 1).
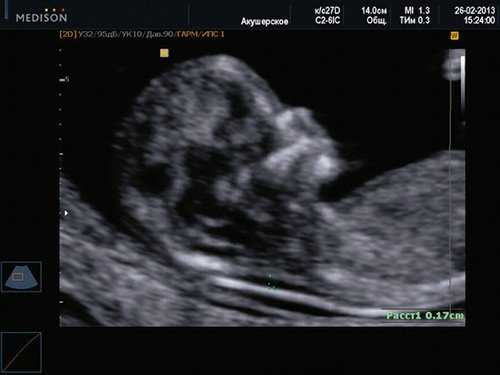
Рис. 1. Нормальная носовая кость.
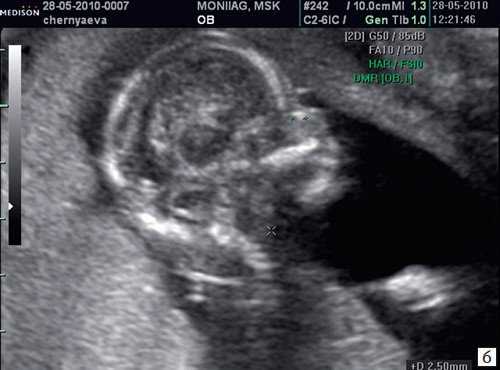
Считается, что носовая кость нормальна, когда она по своей структуре более эхогенна, чем надлежащая кожа и патологична, если она не видна (аплазия) (рис. 2) или ее длина меньше нормы (гипоплазия) (рис. 3). В случае одинаковой или меньшей эхогенности носовой кости чем кожи носовая кость считается патологической (рис. 4).
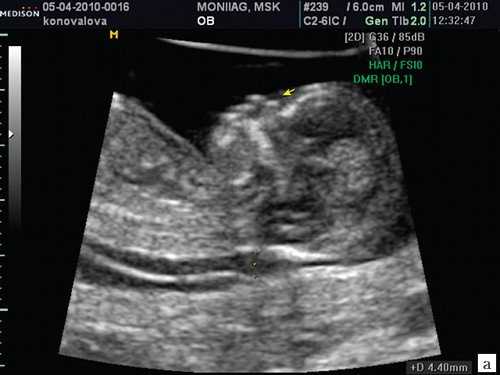
а) Стрелкой указана эхогенная кожа плода.
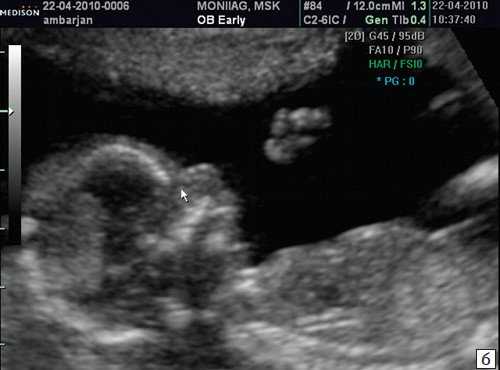
б) Стрелкой указано отсутствие носовой кости.
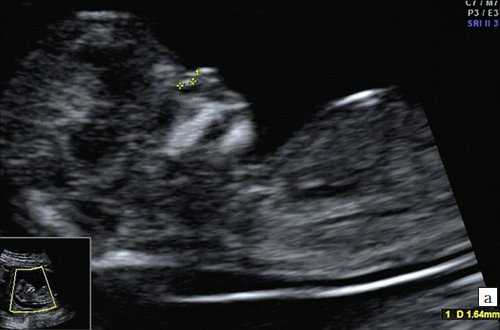
а) Носовая кость в 12 нед и 2 дня длиной 1,4 мм (меньше нижней границы нормы).
б) Носовая кость 2,1 мм в 14 нед у плода с синдромом Дауна.
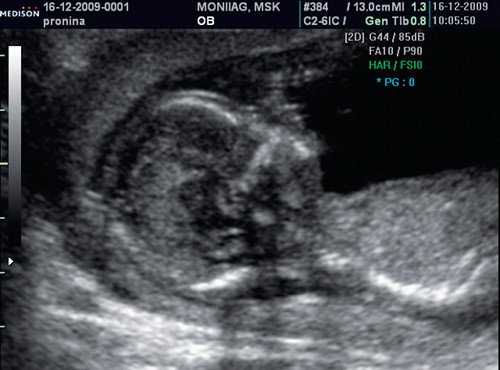
Рис. 4. Сниженная эхогенность носовой кости.
Итак, патологией носовой кости считается:
- отсутствие носовой кости (аплазия);
- изменение ее длины (гипоплазия);
- изменение ее эхогенности.
Учитывая то, что многие работы по изучению этого важного маркера были проведены на различных по составу группах населения, данные по частоте отсутствия носовой кости у разных авторов отличаются. Так, по усредненным данным по мультицентровым исследованиям FMF в 11-14 нед носовая кость отсутствует у эуплоидов (в случае нормального кариотипа) от 1 до 2,6% плодов [2, 5, 6], при хромосомных патологиях: у плодов с трисомией 21 - в 60%, с трисомией 18 - в 50%, у плодов с трисомией 13 - у 40% [3].
Эволюция развития оценки этого маркера и мнение специалистов на этот счет, пожалуй, одна из самых дискутабельных проблем, не до конца решенных в скрининге I триместра беременности. Большинство авторов считают оценку носовой кости в I триместре одной из самых сложных задач среди всех остальных маркеров. И это мнение не лишено оснований.
Безусловно, сторонники теории о том, что для каждой расы (азиаты, афро-американцы и т.д.) и популяции народов (буряты, калмыки, народы Северного Кавказа) должны существовать свои процентильные нормативы для каждого КТР правы. Однако проведение этих исследований возможно лишь тогда, когда в рамках безвыборочного скрининга на нормальных плодах будут проведены мультицентровые исследования с измерением носовой кости.
В программе расчета риска Astraia при оценке носовой кости есть 4 поля: норма, патология (аплазия/гипоплазия), четко не видна, оценить не удалось, т.е. для того, чтобы поставить диагноз "Гипоплазия носовой кости" нужно удостовериться, что она на самом деле меньше нормативных значений для данного срока беременности, а это можно сделать только путем ее измерения и сравнения с известным нормативом.
Метод оценки носовой кости только лишь "да/нет", когда предлагается только увидеть носовую кость и сравнить ее эхогенность с кожей весьма "аппаратозависим", т.е. очень вариабелен и зависит от технических настроек ультразвукового сканера. При получении "жесткого" изображения, характерного для некоторых ультразвуковых аппаратов со специфическими заводскими пресетами (настройками) для осмотра плода в I триместре, всегда эхогенность кожи будет сопоставима, т. е. одинакова с эхогенностью носовой кости. Таким образом, у врачей практического звена, не имеющих возможности работать на сканерах премиум класса, возникают объективные трудности с оценкой этого важного дополнительного диагностического маркера.
Как сторонники метода измерения носовой кости в 11-14 нед приведем данные по Московской области. Область является разнородной по населяющему ее национальному составу. В своей работе мы пользовались нормативными значениями длины носовой кости, опубликованными J. Sonek и соавт. в 2003 году [8], за нижнюю границу нормы принимая значение 5-го процентиля (таблица).
Экспертами окружных кабинетов Московской области проводилась оценка не только присутствия и отсутствия носовой кости, но и ее измерение у всех беременных женщин (около 150 тысяч обследованных за 3,5 года работы скрининга). Все 31 эксперт Московской области имеют действующий сертификат компетенции FMF как на ТВП, так и на оценку носовой кости. Проведенный анализ выявления патологии (аплазия/гипоплазия) носовой кости у плодов с хромосомной патологией показал, что из пренатально выявленных 266 случаев синдрома Дауна у плода в I триместре носовая кость была патологична в 248 случаях, что составляет 93,2%.
Это высокая частота патологии носовой кости при синдроме Дауна свидетельствует о правильно выбранном алгоритме оценки носовой кости от которого мы никогда не намерены отказываться, получая такие высокочувствительные результаты, особенно, что касается диагностики синдрома Дауна. В случаях выявления других хромасомных аномалий, частота выявления патологии носовой кости была сопоставима с данными литературы. При синдроме Эдвардса носовая кость патологична у 78 плодов, что составляет 71%, при синдроме Патау - у 24 (59%) плодов, при моносомии Х - в 24 (42%) случаях, при триплоидии - у 22 (49%) плодов.
Особо хотелось бы подчеркнуть, что в нашем исследовании было 10 беременных корейской национальности, попавших в группу риска по хромосомной патологии. У 4 из них была диагностирована патология носовой кости у плода. Можно было ожидать, что это этническая особенность, однако все данные плоды при пренатальном кариотипировании имели хромосомную патологию (трисомию 21). И, наоборот, у 6 плодов, имеющих нормальный кариотип как по длине, так и по эхогенности носовой кости были в пределах нормативных для данного срока значений.
В работах некоторых авторов установлено, что при трисомии 21 в I триместре беременности лишь у 25% плодов носовая кость отсутствовала, в более высокой частоте она была гипоплазирована (36%) [11].
Так как у нормальных плодов отсутствие носовой кости более характерно для срока 11 нед беременности, чем 13 нед, FMF дает практическую рекомендацию о том, что если в этот срок (11 - начало 12 нед) у плода отсутствует носовая кость при условии нормальных показателей других маркеров (ультразвуковых и биохимических) не стоит учитывать этот показатель при расчете индивидуального риска. В дальнейшем рекомендуется провести дополнительное ультразвуковое исследование через одну неделю. В том случае, если носовая кость останется патологична, необходимо учитывать этот факт при перерасчете величины индивидуального риска по хромосомным аномалиям [4].
Оценка носовой кости улучшает результаты комбинированного скрининга. Частота обнаружения патологии увеличивается с 90 до 93%. Частота ложноположительных результатов уменьшается с 3,0 до 2,5% [2, 3, 5, 6].
Таким образом, собственные данные позволяют нам рекомендовать оценивать носовую кость в сроки 11-14 нед по двум параметрам: эхогенность и длина, принимая за патологию носовой кости ее отсутствие, гипоплазию и снижение эхогенности.
Литература
- Баранов В.С., Кузнецова Т.В., Кащеева Т.К. и др. Современные алгоритмы и новые возможности пренатальной диагностики наследственных и врожденных заболеваний. Методические рекомендации. С.-Петербург, 2013. С. 23-46.
- Nicolaides K.H. Screening for fetal aneuploidies at 11-13 weeks//Prenatal diagnosis. 2011, 31: 7-15.
- Nicolaides K.H. Пер. с англ. Михайлова А., Некрасовой Е. Ультразвуковое исследование в 11-13+6 недель беременности. С.-Петербург, 2007. ИД "Петрополис", 142 с.
- Kagan K.O., Cicero S., Staboulidou I., Wright D., Nicolaides K.H. Fetal nasal bone in screening for trisomies 21, 18 and 13 and Turner syndrome at 11-13 weeks of gestation // Ultrasound Obstet Gynecol. 2009; 33: 259-264.
- Kagan K.O., Staboulidou I., Cruz J., Wright D., Nicoladides K.H. Two-stage first-trimester screening for trisomy 21 by ultrasound assessment and biochemical testing // Ultrasound Obstet Gynecol. 2010. V. 36. N 5. P. 542-547.
- Cicero S., Curcio P., Papageorghiou A., Sonek J., Nicolaides K. Absence of nasal bone in fetuses with trisomy 21 at 11-14 weeks of gestation: an observational study // Lancet 2001; 358:1665-1667.
- Sonek J.D., Mckenna D.,Webb D.,Croom C., Nicolaides K. Nasal bone length throughout gestation: normal ranges based on 3537 fetal ultrasound measurements // Ultrasound in Obstetrics & Gynecology. 2003. V. 21. N 2. P. 152-155.
- Kanellopoulos V., Katsetos C., Economides D.L. Examination of fetal nasal bone and repeatability of measurement in early pregnancy // Ultrasound Obstet Gynecol. 2003 Aug;22(2):131-4.
- Cicero S., Bindra R., Rembouskos G., Tripsanas C., Nicolaides K.H. Fetal nasal bone length in chromosomally normal and abnormal fetuses at 11-14 weeks of gestation // Matern Fetal Neonatal Med. 2002; 11: 400-402.
- Keeling J.W., Hansen B.F., Kjaer I. Pattern of malformations in the axial skeleton in human trisomy 21 fetuses // Am J Med Genet. 1997; 68: 466-471.
Читайте также: