Мышечная дистрофия Эмери-Дрейфуса у детей. Диагностика и лечение
Добавил пользователь Alex Обновлено: 21.01.2026
Дистрофия Эмери-Дрейфуса — это мышечная дистрофия, которая наследуется различными способами. Помимо слабости и мышечной атрофии у людей часто возникают проблемы с сердцем, которые могут быть причиной внезапной смерти.
Мышечные дистрофии — это группа наследственных заболеваний мышц, при которых один или несколько генов Гены Гены представляют собой сегменты дезоксирибонуклеиновой кислоты (ДНК) и содержат код для определенного белка, функционирующего в одном или нескольких типах клеток в организме. Хромосомы — это. Прочитайте дополнительные сведенияСимптомы дистрофии Эмери-Дрейфуса
Мышцы становятся слабыми и истощаются (атрофия), начиная с любого возраста до достижения 20 лет. Наиболее часто поражаются следующие мышцы: плечевые мышцы, мышцы голени и сердечная мышца. Мышцы рук и ног стягиваются в постоянное согнутое положение, которое называется контрактурой. Также может поражаться сердечная мышца (кардиомиопатия Общие сведения о кардиомиопатии Кардиомиопатией называется прогрессирующее нарушение структуры и функции мышечных стенок камер сердца. Различают три основных типа кардиомиопатии: Дилатационная кардиомиопатия, при которой происходит. Прочитайте дополнительные сведенияПрогрессирующая мышечная дистрофия Дрейфуса
Прогрессирующая мышечная дистрофия Дрейфуса — наследственная мышечная миодистрофия, отличающаяся медленным прогрессированием, ранним развитием локтевых контрактур, ретракций шейных мышц и ахилловых сухожилий, сопутствующими нарушениями сердечной проводимости. Заболевание может наследоваться, как сцеплено с Х-хромосомой, так и аутосомно. Мышечная слабость и гипотрофии проявляются преимущественно в мышцах голени и проксимальных отделов рук. Диагностический алгоритм включает исследование неврологического статуса, биохимию крови, ЭМГ и ЭНГ, рентгенографию органов грудной клетки, ЭКГ, эхокардиографию, генеалогический анализ, ДНК-диагностику. Лечение симптоматическое. Могут потребоваться ортопедические операции и имплантация электрокардиостимулятора.
Общие сведения
Мышечная дистрофия Дрейфуса отличается более медленным развитием мышечной слабости, ранним образованием контрактур и сухожильных ретракций, тяжелыми поражениями проводящей системы сердца, требующими имплантации кардиостимулятора. С 1961 года британский невролог Дрейфус и его американский коллега доктор Хоган совместно изучали большую семью, члены которой страдали от миодистрофии, наследуемой сцеплено с Х-хромосомой. Первоначально исследователи считали заболевание доброкачественным клиническим вариантом прогрессирующей миодистрофии Дюшенна, однако в ходе многолетних наблюдений они пришли к выводу о необходимости его выделения в самостоятельную нозологическую единицу. Последующие успехи генетики и биохимии, позволившие установить этиопатогенетический субстрат болезни, подтвердили доводы Дрейфуса и Хогана.
Причины миодистрофии Дрейфуса
Прогрессирующая мышечная дистрофия Дрейфуса является генетически детерминированной патологией и представлена тремя вариантами: рецессивным Х-сцепленным и аутосомными — доминантным и рецессивным. Первый вариант обусловлен мутациями в гене EMD, расположенном на участке Xq26.3 Х-хромосомы и отвечающем за кодирование синтеза белка эмерина. Аутосомные варианты возникают при генетических аберрациях в регионе 1q21.2-q21.3 1-й хромосомы в гене LMNA/C, кодирующем белки ламины A и C. При наличии одной мутации в указанном гене отмечается аутосомно-доминантный тип наследования, а при двух мутациях — аутосомно-рецессивный.
Ламины А/С и эмерин входят в состав мембраны ядерной оболочки миоцитов, поддерживают ее структуру и функции. Недостаток этих белков приводит к нарушению строения мышечных волокон, что наиболее выражено в скелетной и сердечной мышечной ткани. Наблюдения показывают, что при однотипных мутациях мышечная дистрофия Дрейфуса может иметь различные фенотипические проявления, варьирующие от самого мягкого до наиболее тяжелого клинического течения.
Симптомы миодистрофии Дрейфуса
Как правило, мышечная дистрофия Дрейфуса манифестирует в возрасте от 5 до 16 лет. Первыми симптомами заболевания выступают контрактуры локтевых суставов, ретракция (укорочение) мышц заднешейной группы и ахилловых сухожилий. Изменения в шейных мышцах обуславливают вынужденное запрокидывание головы назад и ограничение подвижности позвоночника в шейном отделе. Укорочение ахилловых сухожилий приводит к ходьбе с опорой на наружные края стоп или с постановкой ног на носочки.
Мышечная слабость и гипотрофии обычно появляются позже, вначале малозаметны и характеризуются медленным прогрессированием. В первую очередь поражаются мышцы перонеальной группы (расположенные на внешней стороне голени), мускулатура плечевого пояса и проксимальных отделов верхних конечностей. В отличие от прогрессирующей мышечной дистрофии Беккера, для миодистрофии Дрейфуса не характерны псевдогипертрофии мышц голеней. Мышцы лица остаются интактными. В некоторых случаях наблюдается сколиоз, вызванный ретракцией паравертебральных мышц и контрактурами межпозвоночных суставов. При этом искривление позвоночника носит стабильный характер и не усугубляется со временем.
Развитие детей до возникновения болезни адекватно возрастным нормам. После манифестации миодистрофия медленно прогрессирует вплоть до 20-летнего возраста, когда наблюдается стабилизация процесса. В большинстве случаев пациенты сохраняют способность ходить, подниматься по лестнице, вставать из положения сидя на корточках, не прибегая к приемам Говерса. Главным, отягощающим течение болезни фактором, является патология сердечной мышцы с нарушением сердечной проводимости.
Ранние признаки поражения сердца выявляются лишь при проведении суточного ЭКГ-мониторирования. Приступы брадикардии и синкопальные состояния обычно появляются в возрасте после 20-ти лет, в отдельных случаях предшествуют возникновению мышечной слабости. Могут наблюдаться блокада ножек пучка Гисса, АВ-блокада, предсердные нарушения ритма, дилатационная или гипертрофическая кардиомиопатия. В последующем аритмия может стать причиной инсульта.
Диагностика
Дистрофия Дрейфуса диагностируется совместными усилиями специалистов в области неврологии и генетики. Для выявления сердечной патологии обязательна консультация кардиолога, при наличии деформаций позвоночного столба и контрактур — консультация ортопеда.
Неврологический осмотр определяет умеренное снижение мышечной силы в проксимальных отделах верхних конечностей (бицепс и трицепс плеча, дельтовидная мышца и др.) и дистальных отделах ног (перонеальные мышцы), гипотонию и гипотрофию указанных мышечных групп, сухожильную арефлексию, полную сохранность сенсорной функции. Дополнительно назначаются электронейрография и электромиография, результаты которых указывают на первично-мышечный тип поражения и позволяют исключить другие нервно-мышечные заболевания (миастению, синдром Гийена-Барре, БАС).
В биохимическом анализе крови отмечается умеренное или незначительное повышение уровня КФК (креатинфосфокиназы). Микроскопия образцов, взятых путем биопсии мышц, выявляет наличие гипер- и атрофированных мышечных волокон, отсутствие ядерного окрашивания миоцитов. Характерными ранними изменениями на электрокардиограмме являются снижение амплитуды волны P и удлинение PR-интервала. При отсутствии изменении ЭКГ рекомендовано проведение холтеровского мониторирования. Увеличение границ сердца по данным рентгенографии грудной клетки говорит о наличии кардиомиопатии. Установить более точный кардиологический диагноз позволяет ЭхоКГ.
Окончательный диагноз миодистрофии Дрейфуса может быть подтвержден генетиком по результатам генеалогического анализа и ДНК-диагностики. Дифференциальный диагноз осуществляется с мышечной дистрофией Эрба-Рота, миодистрофиями Дюшенна и Беккета, метаболической миопатией, дерматомиозитом и др. заболеваниями.
Лечение миодистрофии Дрейфуса
Прогрессирующая мышечная дистрофия Дрейфуса, как и другие генные заболевания, не имеет пока эффективного специфического лечения. Проводимая неврологами терапия направлена на поддержание оптимального метаболизма мышц и периферической нервной системы, облегчение нервно-мышечной передачи, предупреждение тромбообразования и церебральной эмболизации, приводящей к инсульту. В рамках этого пациенты получают медикаментозные курсы, включающие назначение АТФ, пентоксифиллина, витаминов В, неостигмина.
Для длительного сохранения больными двигательной функции рекомендовано регулярное проведение массажа и постоянные занятия лечебной физкультурой. В комплексном симптоматическом лечении эффективна физиотерапия — электрофорез неостигмина, тепловые процедуры, водолечение. При образовании контрактур для восстановления способности пациентов самостоятельно двигаться проводят ортопедические операции — тенотомию, тенолиз ахиллова сухожилия, хирургическое устранение локтевых контрактур и т. п.
Ведущую роль в терапии дистрофии Дрейфуса играет кардиологическое лечение. Выявление брадикардии является показанием к имплантации электрокардиостимулятора. При развитии желудочковой аритмии (обычно в позднем периоде заболевания) предпочтительнее установка кардиовертер-дефибриллятора.
Прогноз и профилактика
Прогноз миодистрофии Дрейфуса в основном определяется степенью сердечных нарушений. Зачастую пациенты погибают в среднем возрасте от внезапной сердечной смерти. В других случаях нарастающие сердечные расстройства приводят к декомпенсации с развитием тяжелой сердечной недостаточности, являющейся причиной летального исхода. Иногда смерть пациента связана с инсультом. Профилактические мероприятия включают прохождение консультации генетика перед будущей беременностью, при наличии показаний — проведение пренатальной диагностики с последующим анализом ДНК плода.
Прогрессирующая мышечная дистрофия Беккера
Прогрессирующая мышечная дистрофия Беккера — вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание характеризуется постепенно усугубляющейся и распространяющейся мышечной слабостью, гипотонией и атрофией, первоначально возникающей в мышцах бедер и тазового пояса. Диагностический поиск включает неврологическое обследование, консультацию генетика и кардиолога, нейрофизиологическое тестирование нервно-мышечного аппарата, ДНК диагностику, биопсию мышц с морфологическим, иммунологическим и гистохимическим изучением полученных образцов. Лечение симптоматическое и, к сожалению, малоэффективное. Прогрессирование болезни приводит к потери больными способности самостоятельно передвигаться к возрасту 40 лет.
Прогрессирующая мышечная дистрофия Беккера впервые была описана в 1955 г. как доброкачественный вариант течения мышечной дистрофии Дюшенна. В последующем многочисленные исследования в области клинической неврологии, генетики и биохимии обнаружили существенные отличия в характере течения, биохимической и морфологической основе этих заболеваний. В результате клиническая форма Беккера была выделена как самостоятельная нозология.
Мышечная дистрофия Беккера входит в группу миопатий (миодистрофий) — заболеваний, возникающих вследствие нарушений строения и метаболизма мышечной ткани и проявляющихся мышечной слабостью. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского пола. Частота встречаемости составляет 1 новорожденный на 20 тыс. детей.
Причины
В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30% от общего числа случаев мышечной дистрофии Беккера приходится на т. н. «свежие» мутации. Ген располагается в 21 локусе (в регионе Хр21.2-р21.1) короткого плеча Х-хромосомы. Примерно у 65-70% больных обнаруживаются крупные делеции указанного участка, у 5% - дупликации, у остальных — точковые мутации. Указанные структурные перестройки гена не влекут за собой полного прекращения синтеза дистрофина, как при дистрофии Дюшенна, а потенцируют синтез аномального усеченного белка, в некоторой степени способного выполнять свои функции. Это и обуславливает более доброкачественный характер дистрофии Беккера в сравнении с вариантом Дюшенна.
Патогенез
В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных волокон), обеспечивает эластичность и устойчивость миофибрилл при мышечном сокращении. Неспособность аномального дистрофина адекватно выполнять эти функции приводит к нарушению целостности мембран мышечных волокон. В следствие этого происходят дегенеративные изменения цитоплазматических компонентов последних и повышенная транспортировка ионов калия внутрь миоцитов. Результатом таких биохимических и морфологических сдвигов является гибель миофибрилл и разрушение мышечных волокон. На месте погибших миоцитов происходит образование соединительной ткани, что обуславливает феномен псевдогипертрофии — увеличение объема и плотности мышцы при резком снижении ее сократительной способности.
Симптомы
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в некоторых случаях раньше. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и нижних конечностях. У ряда пациентов первыми проявлениями являются периодические болезненные мышечные судороги (крампи), локализующиеся в ногах. Мышечная слабость обуславливает затруднение при подъеме по лестнице, при необходимости встать из положения сидя. Со временем формируется переваливающаяся «утиная походка». Для того, чтобы встать, пациент вынужден использовать вспомогательные миопатические приемы — опираться руками о расположенные рядом предметы мебели или, при отсутствии таковых, использовать в качестве опоры собственное тело (симптом Говерса).
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В первую очередь поражаются мышцы бедра и тазового пояса, затем процесс распространяется на мускулатуру плечевого пояса и проксимальных мышц рук. В начале болезни формируются псевдогипертрофии, наиболее выраженные в икроножных, дельтовидных, трех- и четырехглавых мышцах. По мере прогрессирования миодистрофии они трансформируются в мышечные гипотрофии.
Клиническая картина мышечной дистрофии Беккера во многом сходна с миодистрофией Дюшенна. Усугубление мышечной слабости с течением времени приводит к обездвиженности пациента и формированию контрактур суставов. Однако развитие дистрофического процесса в мышечной ткани при дистрофии Беккера идет гораздо медленнее, что обуславливает длительную двигательную активность больных. В среднем пациенты сохраняют способность самостоятельно передвигаться до 35-40-летнего возраста. Кроме того, дистрофия Беккера не сопровождается олигофренией, выраженным искривлением позвоночника и другими скелетными деформациями. Возможна кардиомиопатия дилятационного или гипертрофического типа, блокада ножек пучка Гисса, но сердечно-сосудистые расстройства выражены умеренно. Может наблюдаться снижение либидо, гинекомастия, атрофия яичек, импотенция.
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического тестирования. В неврологическом статусе наблюдается снижение мышечной силы и умеренное снижение мышечного тонуса в проксимальных отделах конечностей, выпадение коленных рефлексов при симметричном снижении сухожильных рефлексов дистальных отделов ног и верхних конечностей, полная сохранность чувствительности.
Среди клинических анализов наибольшее значение имеет биохимический анализ крови, который выявляет многократное повышение уровня КФК. Данные электронейрографии позволяют исключить поражение нервных волокон, электромиография свидетельствует о первично-мышечном типе поражения. Биопсия мышц проводится только после отрицательных результатов генетического анализа. Морфологическое исследование полученного материала определяет диффузную разнокалиберность, дистрофические и некротические изменения мышечных волокон, разрастание соединительной ткани. Проводится специальное иммунное окрашивание образцов с последующим определением наличия в них дистрофина.
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или делеций в гене Хр21 дает возможность установить точный диагноз. Отрицательный результат анализа ДНК не говорит об отсутствии патологии, поскольку могут иметь место точковые мутации, поиск которых представляет собой сложную и более дорогостоящую процедуру.
С целью выявления сердечной патологии назначается электрокардиография, Эхо-КГ, консультация кардиолога. Кардиологическое обследование может обнаружить нарушение внутрижелудочковой проводимости, АВ-блокаду, дилатацию желудочков, гипертрофические изменения миокарда, кардиомиопатию, сердечную недостаточность.
Пренатальная диагностика рекомендована, когда мать является носителем патогенного гена. Если ребенок мужского пола, то вероятность развития заболевания у него составляет 50%. Биопсия хориона может проводиться в сроке 11-14 нед. беременности, амниоцентез — после 15-й недели, забор пуповинной крови (кордоцентез) — на сроке больше 18 нед.
Дифференциальная диагностика
Дифференциальная диагностика проводится с прогрессирующей мышечной дистрофией Дрейфуса, миодистрофией Дюшена, мышечной дистрофией Эрба-Рота, метаболической миопатией, полимиозитом и дерматомиозитом, воспалительной миопатией, спинальной амиотрофией, наследственной полиневропатией.
Лечение миодистрофии Беккера
На современном этапе несколькими группами ученых ведутся настойчивые исследования в области поиска эффективных методов лечения прогрессирующих миодистрофий. В настоящее время пациенты получают в основном метаболическую и симптоматическую терапию. Разработаны различные схемы лечения, позволяющие улучшить двигательные возможности больного и несколько замедлить прогрессирование болезни.
Терапия глюкокортикоидами используется для снижения скорости прогрессирование атрофии мышечной ткани. Пациентам назначают метаболические средства (витамины группы В, левокарнитин), витамин D и препараты кальция для профилактики остеопороза, β-адреноблокаторы и ингибиторы АПФ для предупреждения кардиомиопатии, диуретики при сердечной недостаточности.
Наблюдения показали, что постельный режим усугубляет мышечную слабость. Поэтому пациентам рекомендуется умеренная физическая активность, занятия плаванием. Поддержание мышечной эластичности и силы, а также профилактика контрактур проводится средствами массажа, физиотерапии и лечебной гимнастики. Применение различных ортопедических средств (ходунков, инвалидных колясок, фиксаторов для ног, экзоскелетов) позволяет расширить двигательные возможности пациентов и их способность к самообслуживанию. По показаниям проводится хирургическое лечение контрактур.
Прогноз и профилактика
Прогрессирующая мышечная дистрофия Беккера имеет неблагоприятный прогноз. Хотя обездвиженность у пациентов наступает гораздо позже, чем при дистрофии Дюшенна, в конечном итоге поражение сердечной мышцы и дыхательной мускулатуры приводят к гибели пациентов от сердечной или дыхательной недостаточности. Продуманный уход, адекватная терапия, вентиляционная поддержка дыхания, применение ортопедических средств могут лишь увеличить продолжительность и улучшить качество жизни пациента. Профилактика заключается в предупреждении рождения ребенка с патологией путем генетического консультирования будущих родителей и проведение пренатальной диагностики.
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Мышечная дистрофия, или миодистрофия
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Мышечная дистрофия: причины появления, симптомы, диагностика и способы лечения.
Определение
Мышечные дистрофии - это большая группа наследственных заболеваний, которые характеризуются прогрессирующей слабостью и дегенерацией скелетных мышц, то есть потерей мышечной массы. К наиболее распространенным миодистрофиям относятся миодистрофия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия), мышечная дистрофия Дюшенна и дистрофия Беккера. Также в эту группу входят дистрофия Эмери-Дрейфуса, миодистрофия Эрба-Рота и другие.
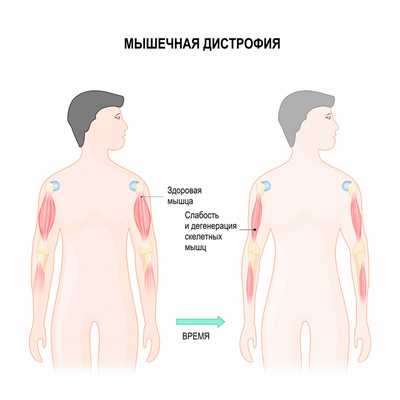
Миодистрофия Дюшенна поражает в основном мальчиков, а распространенность этого заболевания составляет 3,3 на 100 000 населения, 1 из 3500 новорожденных мальчиков страдает данной патологией. Этот вид дистрофии часто выделяют в одну группу с миодистрофией Беккера (частота встречаемости 1 на 20 000 новорожденных). Миодистрофии Дюшенна и Беккера наследуются по Х-сцепленному рецессивному типу. Это означает, что повреждение находится в Х-половой хромосоме и передается от матери к сыну, а дочери являются носительницами и, как правило, сами не болеют.
Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей.
Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них - Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
Частота встречаемости миодистрофии Эрба-Рота составляет от 1,5 до 2,5 случаев на 100 тыс. населения. Этому типу мышечной дистрофии подвержены и мальчики, и девочки. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно, то есть патология проявляется, если ребенок получает аномальный ген от каждого из родителей. Каждый родитель может быть носителем дефектного гена, но обычно остается здоровым. Около 30% генных мутаций возникают de novo, то есть не наследуются, а появляются «ниоткуда» и далее могут передаваться потомству.
Причины появления мышечной дистрофии
Причиной развития разных миодистрофий являются патологии в генах - известно порядка 25 генов, ответственных за развитие врожденных миодистрофий.
При мышечной дистрофии Дюшенна вследствие мутации нарушается выработка белка дистрофина, который обеспечивает прочность, стабильность и функциональность мышечных волокон, и его нехватка приводит к повреждению мембран мышечных клеток (миоцитов).
Классификация заболеваний
Мышечные дистрофии могут классифицироваться в зависимости от того, какой белок подвергся мутации. Кроме того, их подразделяют по типу наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные.
Симптомы мышечной дистрофии
Мышечная дистрофия Дюшенна обычно манифестирует в возрасте 2-3 лет. Патологические процессы сначала происходят в мышцах ног, дети могут ходить на пальцах, вразвалку, отмечается избыточное выгибание позвоночника вперед - лордоз. Детям становится сложно бегать, прыгать, подниматься по лестнице, вставать с пола. Для разных типов миодистрофий характерным является симптом Говерса - вследствие слабости мышц бедер и тазового пояса больному, чтобы подняться из положения на корточках, приходится опираться руками об пол, затем подниматься, опираясь руками об колени. Мышечная слабость прогрессирует, у детей развивается сколиоз и сгибательные контрактуры - когда ребенок не может полностью разогнуть конечность. Дети часто падают, поэтому велик риск переломов рук или ног. Отдельные мышечные группы могут замещаться жировой или фиброзной тканью, в результате чего появляется псевдогипертрофия мышц, особенно заметная на лодыжках. Если страдает миокард (сердечная мышца), то существует предрасположенность к развитию нарушений ритма и проводимости сердца, а также дилатационной кардимиопатии (состояния, когда камеры сердца увеличены, а стенки истончены), приводящей к сердечной недостаточности.
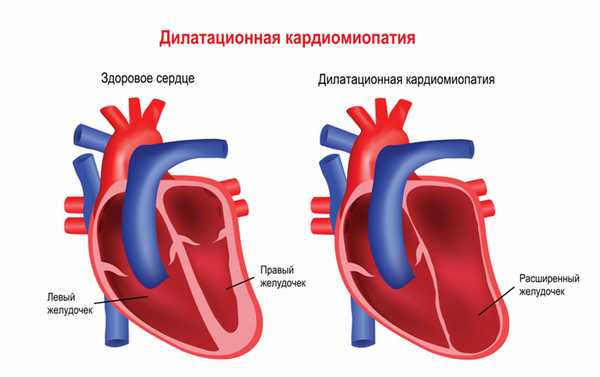
В 20-30% случаев при мышечной дистрофии Дюшенна появляются нарушения интеллекта и памяти.
К 12 годам большинство детей вынуждено пользоваться инвалидной коляской. В возрасте 15-20 лет пациентам уже требуется респираторная поддержка, умирают больные миодистрофией Дюшенна от дыхательных или кардиальных осложнений в возрасте 12-25 лет.
Миодистрофия Беккера дебютирует в 10-20 лет и медленно прогрессирует, способность к самостоятельной ходьбе сохраняется в течение 15-20 лет от начала заболевания. Симптоматика схожа с миодистрофией Дюшенна, слабость распространяется на мышцы бедер, таза, плеч, пациенты ходят на носочках или вразвалку, также наблюдается гипертрофия мышц голеней.
Развитие заболевания очень индивидуально, некоторым пациентам требуется инвалидное кресло к 30 годам, некоторые длительное время обходятся тростью.
У пациентов с миодистрофией Беккера также отмечается поражение сердечной мышцы с развитием сердечной недостаточности.
Первые признаки миодистрофии Ландузи-Дежерина проявляются в основном в возрасте 10-20 лет. Сначала атрофия и мышечная слабость наблюдаются в плечевом поясе с поражением мышц лопаток и плеч, потом распространяются на лицо с характерной асимметричностью. Начальными проявлениями являются затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании заболевания страдают лицевые мышцы, при этом пациент не может крепко зажмурить глаза и сжать губы. Позже мимика становится скудной, а речь неразборчивой. Характерными симптомами являются поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира»), «полированный» лоб. Иногда атрофия распространяется на мышцы ног. Другими клиническими признаками миодистрофии Ландузи-Дежерина могут быть аномалии сосудов сетчатки глаза, отек и отслойка сетчатки, снижение слуха.
Для миодистрофии Эмери-Дрейфуса характерны контрактуры локтевых и голеностопных суставов, возникающие в раннем детстве (укорочение ахилловых сухожилий приводит к тому, что ребенок не может опуститься на пятки), тугоподвижность позвоночника, медленно прогрессирующая слабость лопаточно-плечевых и тазово-перонеальных мышц (мышц бедра и голени), а также выраженная кардиомиопатия с нарушениями ритма и проводимости. Тяжесть заболеваний сердца часто определяет прогноз течения болезни вследствие высокой вероятности внезапной сердечной смерти или развития прогрессирующей сердечной недостаточности.
Миодистрофия Эрба-Рота сопровождается слабостью мышц поясничной области и конечностей. Первые признаки появляются в возрасте 10-20 лет: трудности при беге, быстрой ходьбе, прыжках, характерен симптом Говерса. Со временем начинает меняться осанка, походка, снижается тонус мышц плечевого пояса. С прогрессированием заболевания больной может полностью потерять способность ходить. Тотальная гипотрофия мышц туловища приводит к тому, что у пациента начинают выступать лопатки, талия становится очень тонкой, усиливается поясничный лордоз. Характерен симптом свободных надплечий — при попытке приподнять больного, удерживая его подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними.
Диагностика мышечной дистрофии
Предварительный диагноз врач может установить уже при осмотре, наблюдая за попытками ребенка побежать, прыгнуть, подняться по ступенькам, встать с пола.
Для подтверждения диагноза проводятся:
- Анализ крови на сывороточную креатинкиназу, АСТ, АЛТ.
Читайте также:
- Относительное расположение локтевой кости
- Лечение пароксизмальных наджелудочковых тахикардий (ПНЖТ) с механизмом re-entry. Критерии диагностики
- Хирургическая анатомия внутреннего слухового канала
- Андрогены. Регуляция секреции и физиологические эффекты половых стероидов коры надпочечников. Вирилизация.
- Общие сведения о синдромах хромосомной делеции