УЗИ, рентгенограмма при гипофасфатазии у плода
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Детская форма гипофосфатазии в реальной клинической практике
Гипофосфатазия (ГФФ) — редкое врожденное рахитоподобное заболевание, обусловленное снижением активности тканенеспецифической щелочной фосфатазы (ЩФ). Для ГФФ характерны нарушение минерализации скелета, костные деформации, выпадение зубов в раннем детском возрасте, дыхательные расстройства и неврологические нарушения. В зависимости от времени манифестации заболевания выделяют перинатальную (первые проявления во внутриутробном периоде или при рождении), инфантильную (манифестация до 6 мес.), детскую (с 6 мес. до 18 лет) и взрослую (18 лет и старше) формы ГФФ. Степень тяжести проявлений обычно обратно пропорциональна возрасту дебюта болезни.
Целью работы стало описание клинического случая ГФФ, манифестировавшего в детском возрасте, и выявление клинических симптомов, наиболее типичных для детской формы ГФФ.
Описание клинического случая
Пациент — мальчик 2016 года рождения, от беременности, протекавшей на фоне хронической внутриутробной гипоксии. Появился на свет после срочных самостоятельных родов с нормальными массо-ростовыми показателями (3800 г, 52 см), оценка по шкале Апгар — 8/9 баллов. На грудном вскармливании до 3 месяцев. Профилактические прививки по национальному календарю, без реакций. Травм и операций не было. Аллергоанамнез не отягощен. Родители среднего роста (мать — 160 см, отец — 170 см). Эндокринные заболевания у ближайших родственников не выявлены.
Ребенок с рождения наблюдался ортопедом по поводу врожденной левосторонней косолапости, в связи с чем проводилось консервативное лечение с положительной динамикой. Кроме этого, пациент был неоднократно осмотрен неврологом по поводу диффузной мышечной гипотонии, на фоне которой отмечалась задержка моторного развития. На первом году жизни также обращали на себя внимание частые респираторные инфекции и бронхиты, в 1 год 1 месяц ребенок перенес пневмонию.
В 2,5 года ребенка впервые проконсультировал детский эндокринолог в Детской городской поликлинике № 6 г. Калининграда, выявлено снижение уровня ЩФ, и по совокупности клинических, рентгенологических и лабораторных данных диагностирована ГФФ. В дальнейшем диагноз был подтвержден молекулярно-генетически: в гене ALPL найдена компаунд-гетерозиготная мутация с.571 G>A/с.144_148dup.
Повторное обследование, проведенное в детском эндокринологическом отделении Университетской детской клинической больницы ФГАОУ ВО «Первый МГМУ им. И.М. Сеченова» в возрасте 3,5 года, подтвердило выраженную задержку роста — рост 87 см соответствует -2,55 стандартного отклонения (SD), масса тела соответствует верхней границе нормы — 14 кг (ИМТ — 18,5 кг/м 2 или +2,04 SD). При осмотре обращает на себя внимание отсутствие 7 зубов.
Заключение.
Характерными проявлениями детской формы гипофосфатазии являются задержка роста и мышечная гипотония, которая может приводить к задержке моторного развития и нарушению походки; боли в мышцах и выраженная утомляемость, ограничивающие время ходьбы; рахитические деформации, в том числе вальгусные деформации нижних конечностей; раннее выпадение молочных зубов с неизмененным корнем, дыхательные расстройства и частые бронхолегочные заболевания.
Диагноз может быть установлен на основании данных анамнеза, характерной клинической картины, результатов рентгенологического и лабораторного исследований (выраженное снижение уровня щелочной фосфотазы (ЩФ) и подтвержден данными молекулярно-генетического исследования.
Тяжесть состояния пациента в описанном клиническом случае обусловлена снижением уровня ЩФ, олигодонтией (с момента манифестации заболевания выпали 7 молочных зубов), задержкой роста, выраженными рахитическими деформациями грудной клетки, которые повышают риск рецидивирующих респираторных заболеваний; деформацией нижних конечностей, мышечной гипотонией, ограничивающими возможность самостоятельного передвижения ребенка (нарушение походки по парапаретическому типу, самостоятельно проходит не более 10 метров), тяжелым нарушением минерализации костной ткани, согласно рентгенографии.
Опираясь на данные литературы, можно предположить, что назначение патогенетической фермент-заместительной терапии препаратом асфотазы альфа позволит улучшить качество жизни ребенка благодаря регрессу симптомов заболевания.
Случай диагностики одонтогенной формы гипофосфатазии у трёхлетнего ребенка
22 декабря 2017 года через соцсети ко мне обратилась женщина с жалобами, что у её трехлетнего сына выпадают здоровые молочные зубы с нерассосавшимися корнями.
Жалобы
У ребенка с 2 лет наблюдалось постепенное выпадение молочных зубов. К трём годам отсутствовало 5 зубов. Выпадали безболезненно, предварительно расшатываясь. У пациента наблюдалось килевидное искривление грудной клетки, общая слабость.
Зубы выпадали без внешнего воздействия. Ортодонты явных причин для ранней потери зубов не выявили, коррекция не была предложена, учитывая возраст пациента.
Анамнез
Ребёнок от первой беременности,протекавшей с угрозой, первых срочных родов. Вес при рождении — 3250 гр, рост — 51 см. По шкале Апгар — 7/8 баллов, трёхкратное обвитие пуповины вокруг шеи. Выписан с мамой на 5 сутки. С рождения наблюдается у невролога с диагнозом "гипотонус конечностей", проходит курсы массажа, ЛФК. Наблюдается умеренная гидроцефалия, лечение не требуется. Хирург на плановом медосмотре зарегистрировал искривление грудной клетки. Ребёнок пониженного питания. Рост и развитие соответствуют возрасту. Педиатр предположил диагноз "рахит", начато соответствующее лечение, без эффекта. При проведении онлайн-консультации заподозрен диагноз "гипофосфатазия, детская форма".
Обследование
Ребёнок пониженного питания, астенического типа телосложения. Вес 12,500 кг, рост 94 см. Голова гидроцефальной формы с выступающими лобными буграми, зубы с жёлтой эмалью. Имеется умеренная килевидная деформация грудной клетки, выступающие рёберные дуги, клинодактилия мизинцев, плосковальгусные стопы. Отмечается отсутствие 5 молочных зубов. Зона суставов не изменена. Патологии со стороны дыхательной, пищеварительной,нервной, сердечно-сосудистой систем не выявлено.
Данные лабораторной диагностики: — щелочная фосфатаза 85 (снижение); — кальций 2,46; — фосфор 2,10; — общий анализ крови без патологии; — общий анализ мочи без патологии; — Кальций в моче 3,12 ммоль/л. Проведено исследование щитовидной железы-без патологии. Рентгенография кисти — костный возраст соответствует 1,5-2 годам. На рентгенографии грудной клетки выявлена умеренная деформация, не требующая коррекции. На основании значительного снижения уровня щелочной фосфатазы ребёнок был направлен в московский Центр Молекулярной генетики, где было проведено исследование мутаций в гене ALPL и выявлены мутации в 5 и 6 экзонах. На основании молекулярно-генетического анализа диагноз "гипофосфатазия" подтверждён. Обследование родителей показало, что отец и мать являются носителями мутаций в гене ALPL в гетерозиготном состоянии. У родного брата также выявлена одна мутация в гене, ответственном за развитие гипофосфатазии в гетерозиготном состоянии.
Диагноз
Лечение
Лечение данного заболевания финансируется из средств бюджета, так как гипофосфатазия относится к орфанным заболеваниям. Лечение показано при жизнеугрожающих и инвалидизирующих состояниях. В данном случае оно не показано.
Заключение
Гипофосфатазия — это наследственное заболевание, частота которого составляет 1:100000, однако в России выявлено только 19 человек с данным заболеванием, что говорит о малой осведомлённости врачей о данной патологии, сложностях диагностики, дифференциации с другими заболеваниями и реабилитации. Лечение возможно в НИИ педиатрии в г. Москве. Следует помнить, что пациентам с данной патологией противопоказан приём витамина Д и препаратов кальция, обычно применяющихся для лечения рахита, заболевания со схожей симптоматикой, так как высокие дозы кальция могут усилить проявления основного заболевания, привести к тяжёлой гиперкальциемии.
Гипофосфатазия - симптомы и лечение
Что такое гипофосфатазия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 7 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
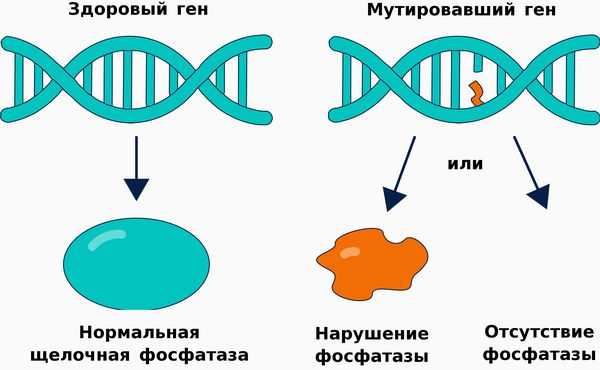
Гипофосфатазия — это наследственное метаболическое заболевание, связанное со снижением активности либо с полным отсутствием тканенеспецифической щелочной фосфатазы. Эта редкая болезнь затрагивает многие органы и системы и является жизнеугрожающей для пациента. [20] [23]
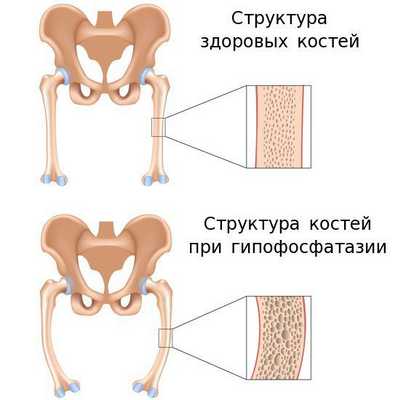
Такой фермент, как щелочная фосфатаза, играет важную роль в минерализации тканей. Поэтому её дефицит становится причиной различных системных нарушений, проявляющихся недостаточной минерализацией костей и их деформацией. При тяжёлых формах гипофосфатазии у новорождённых также развивается дыхательная недостаточность и судороги.
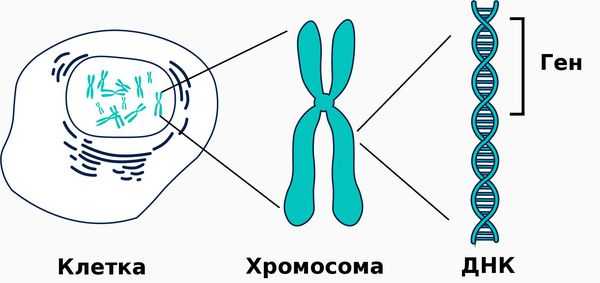
Причиной заболевания является мутация в гене ALPL, кодирующем указанный фермент. Этот ген расположен на коротком плече первой хромосомы, которая содержит большее количество генетической информации о структуре организма человека по сравнению с остальными хромосомами.
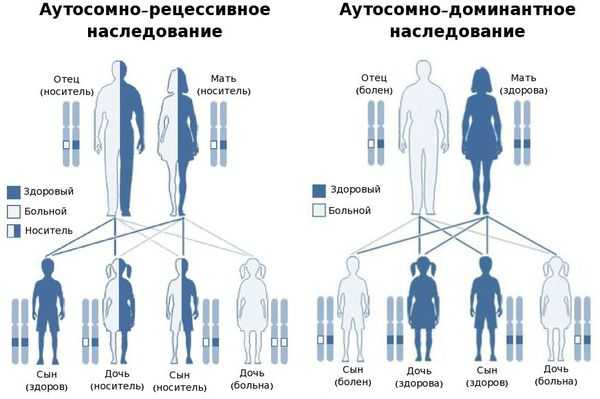
В большинстве случаев заболевание передаётся по аутосомно-рецессивному типу, т. е. при условии, что мутировавший ген передался к ребёнку от обоих родителей. Также описаны и случаи аутосомно-доминантного типа наследования — передача мутировавшего гена только от одного родителя. [25]
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы гипофосфатазии
Клиническая картина заболевания в значительной степени зависит от возраста пациента и формы заболевания. [1]
К основным проявлениям гипофосфатазии относят повреждения скелета в виде:
- значительной гипоминерализации костей — нарушения развития костной ткани;
- краниосиностоза — деформации черепа в связи с преждевременным закрытием черепных швов;
- рахитоподобной деформации скелета;
- остеомаляции — ухудшение прочности костей в связи с их недостаточной минирализацией;
- незаживающих переломов;
- повторяющихся переломов, требующих применения инвалидного кресла и других способов поддержки для передвижения. [23]
Боли в суставах, мышцах и костях — одно из значительных и прогрессирующих проявлений гипофосфатазии. [14]
Поражение дыхательной системы проявляется гипоплазией (недоразвитием) лёгких и дыхательной недостаточностью, при которой в некоторых случаях требуется применение кислородной поддержки. [23]
Со стороны центральной нервной системы наблюдаются судорожные приступы, повышение внутричерепного давления, различные внутричерепные кровотечения. [20]
Очень часто при гипофосфатазии встречаются тяжёлые поражения почек в виде нефрокальциноза и почечной недостаточности.
Патология мышечной системы проявляется различными миопатиями, хронической болью в мышцах, задержкой или полным отсутствием моторного развития. [22]
Характерны поражения суставов в форме хондрокальциноза (псевдоподагры) и прогрессирующего артрита.
Гипоминерализация костей приводит к раннему выпадению зубов у детей и потере здоровых зубов у взрослых. [20]
Ещё одним характерным проявлением гипофосфатазии является отставание костного возраста от паспортного по результатам рентгенологического исследования.
При перинатальной форме гипофосфатазии отмечается укорочение трубчатых костей, недоразвитие и укорочение рёбер, мягкие кости черепа, повышается вероятность поражения центральной нервной системы в процессе родов. [20]
Патогенез гипофосфатазии
Патогенез гипофосфатазии связан со снижением выработки либо активности щелочной фосфатазы, структура и функционирование которой нарушается п ри наличии мутации в гене ALPL .
Щелочная фосфатаза принимает участие в метаболизме трёх важных субстратов:
- пиридоксаль-5-фосфат (витамин В6 , PLP);
- неорганический пирофосфат (PPi);
- фосфоэтаноламин (PEA).
Нарушение метаболизма пиридоксаль-5-фосфата приводит к повышению его циркуляции в крови и недостатку активной формы витамина В6. В циркулирующей форме витамин В6 не может проникнуть через гемато-энцефалический барьер. Недостаток активной формы витамина сопровождается поражением центральной нервной системы, что проявляется развитием судорожного синдрома, снижением мозговой активности и трудностями в усвоении информации. [23]
Неорганический пирофосфат является основным компонентом гидроксиапатита, входящего в состав костей. При нарушении метаболизма неорганического пирофосфата происходит повышение его концентрации в крови, гиперфосфатемия и гиперкальциемия, что приводит к отложению солей кальция в суставах, почках и других внутренних органах:
- отложение солей кальция в суставах является причиной артритов, в том числе, тяжёлых;
- поражение почек приводит к развитию почечной недостаточности и полиорганным нарушениям.
Неорганический пирофосфат накапливается и в швах черепа, что сопровождается развитием краниосиностоза, нарушением роста головного мозга, родовым травматизмом, внутричерепной гипертензией, мозговыми кровоизлияниями и отёком диска зрительного нерва. [23]
В результате этих процессов снижается содержание гидроксиапатита в костях, что приводит к деминерализации костей, их искривлению, укорочению и переломам. Патология цементирования зубов является причиной их раннего выпадения. [16] Из-за деформации грудной клетки развивается дыхательная недостаточность и гипоплазия лёгких. Снижение плотности и прочности костей черепа приводит к травмам головного мозга. Искривления позвоночника вызывают сдавление спинного мозга и нарушение осанки. [22]
Фосфоэтаноламин является промежуточным метаболитом, нарушение его метаболизма сказывается на различных реакциях.
Классификация и стадии развития гипофосфатазии
Выделяют четыре формы гипофосфатазии:
- пренатальная (предродовая);
- инфантильная;
- детская;
- взрослая.
При пренатальной форме клинические проявления развиваются ещё до родов и характеризуется задержкой внутриутробного роста плода, гипоксией, дефектами черепа ("мембранозный череп"), деформациями костей, тяжёлой патологией грудной клетки и гипоплазией лёгких. Такие дети в большинстве случаев погибают внутриутробно либо рождаются преждевременно, у них развивается дистресс-синдром (тяжёлое расстройство дыхания). [20] Ранее считалось, что 100% таких детей погибает, но в последнее время благодаря своевременной диагностике и лечению возможно выхаживание пациентов с пренатальной формой гипофосфатазии и продление их жизни. Данную форму заболевания можно диагностиравать во время ультразвукового исследования в период беременности по характерным эхографическим признакам. Тип наследования: аутосомно-рецессивный. [25]
Инфантильная форма проявляется в скором времени после рождения. Смертность таких пациентов без лечения достигает 40%. В неонатальном периоде дети с этой формой гипофосфатазии возбудимы, им характерен плохой аппетит, наблюдается повышенная реакция на внешние раздражители, судороги, диспепсия (нарушение пищеварения), обезвоживание, снижение мышечного тонуса, тяжёлые костные аномалии (мягкие кости, увеличенные черепные швы и роднички), повышение кальция в крови. Часто смерть наступает в неонатальном периоде в связи с тяжёлой дыхательной недостаточностью, но известны случаи спонтанного улучшения. [17] Деформация грудной клетки в 50% случаев приводит к развитию пневмонии . В 64% случаев пациентам требуется кислородная поддержка, 66% детей с этим заболеванием нуждаются в искусственной вентиляции лёгких, из которых 95%, к сожалению, умирают. [17] Тип наследования: аутосомно-рецессивный. [25]
При детской форме гипофосфатазии наблюдается постепенное развитие рахитоподобных скелетных изменений, ранняя потеря зубов, хондрокальциноз и артропатии. При данной форме заболевания очень часто ошибочно ставится диагноз "Рахит", что приводит к неправильному лечению и развитию осложнений. Тип наследования: аутосомно-рецессивный. [25]
При взрослой форме наблюдаются частые переломы и снижение минеральной плотности костей, [7] выпадение зубов, искривление конечностей, встречается изолированная фосфоэтаноламинурия. При рентгенографии выявляется уменьшение минеральной плотности костей черепа, трубчатых костей и отложение кальция в почках. Тип наследования: аутосомно-доминантный. [25]
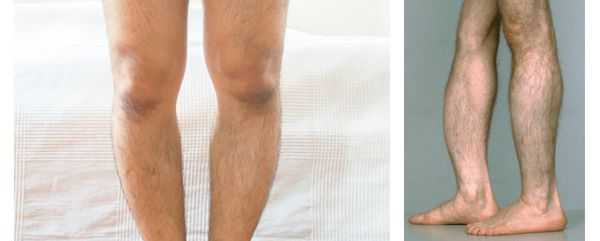
Некоторые авторы выделяют отдельно зубную форму гипофосфатазии . Она характеризуется выпадением здоровых зубов и отсутствием другой симптоматики. Считается самым лёгким вариантом течения гипофосфатазии. [22]
Осложнения гипофосфатазии
Гипофосфатазия является жизнеугрожающим состоянием.
В раннем детском возрасте основными осложнениями заболевания, приводящими к смерти, являются дыхательная недостаточность, травмы головного мозга, внутримозговые кровоизлияния. [12]
В более старшем возрасте развивается почечная недостаточность, связанная с нефрокальцинозом (отложением солей кальция в почках). Это приводит к полиорганной недостаточности и смерти.
Инвалидизация пациентов происходит за счёт снижения мышечного тонуса и артропатий. Поэтому многие пациенты нуждаются в специальных устройствах для облегчения передвижения, в том числе, в инвалидных креслах. [22]

Недостаток биодоступного витамина В6 вызывает судороги, снижение когнитивных функций, задержку психического развития. [8] Эти осложнения лечат витаминами группы В, однако это приводит к избытку пиридоксаль-5-фосфата в крови.
Краниосиностоз приводит к нарушению роста головного мозга, повышению внутричерепного давления, многим пациентам требуется оперативное лечение.
Очень часто осложнения наступают по причине неправильно поставленного диагноза и соответствующего лечения. Так, детям с гипофосфатазией во многих случаях ошибочно ставится диагноз "Рахит" и проводится лечение витамином Д и препаратами кальция. Это приводит к гиперкальциемии, усиленному отложению солей в суставах и почках, а также к гипервитаминозу Д.
Диагностика гипофосфатазии
Диагностика гипофосфатазии основывается на выявлении снижения щелочной фосфатазы в крови.
Диагностика пренатальной и инфантильной гипофосфатазии
Диагностировать пренатальную форму гипофосфатазии возможно во время беременности. Ей характерны следующие ультразвуковые признаки:
- отсутствие оссификации костей (их окостенения);
- более чёткая визуализация структур головного мозга;
- положительный "тест надавливания"; [5]
- недостаточная оссификация, укорочение и деформация длинных трубчатых костей;
- специфическая картина метафиза (участка трубчатой кости);
- характерные акустические дорожки на ультразвуковом снимке. [22][24]
Отсутствие оссификации костей черепа и "мембранозный череп" являются проявлениями, общими для несовершенного остеогенеза и гипофосфатазии, поэтому необходима дифференциальная диагностика этих состояний. [17]
При ультразвуковом исследовании , как правило, трудно дать количественную оценку остеогенеза. Однако если на картине УЗИ внутренняя структура черепной коробки и поверхность мозга, которые обычно невидны у нормально развивающегося плода, визуализируются ярче обычного, то это можно принять в качестве особенности нарушенной формирования костей черепа. [24]
Далее, если провести "тест надавливания" — сильно нажать датчиком ультразвука на брюшную стенку матери — и посмотреть, насколько деформируется череп плода, то у нормально развивающегося малыша форма черепа сохранится (так как кости черепа твёрдые), а в случае выявленного нарушенного остеогенеза костей черепа и "мембранозного черепа" будет наблюдаться его деформация. [24]
Обычно считается, что для того, чтобы увидеть состояние костей, лучше прибегнуть не к ультразвуковой, а к рентгенологической диагностике . На рентгенограмме ясно видны укорочение и искривление длинных трубчатых костей, а также просветление "языков", которые выступают из метафиза в сторону диафиза, являющееся наиболее специфической диагностической картиной при гипофосфатазии. [24]
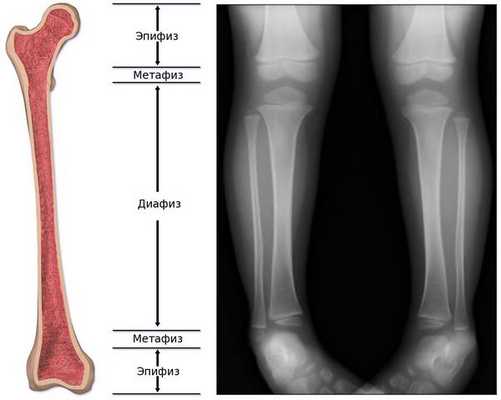
С другой стороны, на ультразвуковом снимке можно убедиться в таких особенностях, как укорочение длинных трубчатых костей, загибы и деформация, но при этом трудно определить, является ли искривление следствием перелома кости или изгиба, а также трудно визуализировать просветление пониженной плотности ("языки пламени" у более взрослых пациентов). [24]
При гипофосфатазии происходит относительно хорошее окостенение средней части диафиза (тела) длинных трубчатых костей, однако к периферии костей оссификация постепенно ухудшается, и на участках метафиза видна только поверхность костей. Поэтому на рентгенограмме это визуализируется как просветление "языков" пониженной плотности. [13]
На ультразвуковом снимке относительно окостеневшие средняя и периферическая части диафиза визуализируются как линии низкой плотности. [10] С другой стороны, рядом с метафизом на поверхности наблюдается незначительная оссификация, изображение поверхности кости нечёткое, под диафизом с обеих сторон появляются "пояски". Всё это является специфической ультразвуковой картиной недостаточно "окрепших" костей при гипофосфатазии и не наблюдается при нормальном развитии плода. [24]
Описание характерных акустических дорожек
Ультразвуковой луч, прошедший через мягкие ткани и достигший поверхности кости, на 90% отражается от её поверхности, что отображается на ультразвуковом снимке в качестве линии низкой эхогенности (плотности). Однако в перинатальном периоде эпифиз практически не заметен и даже у нормально развивающегося плода не визуализируется на ультразвуковом снимке.
В основном у льтразвуковой луч отражается от поверхности кости, а сквозь кость проходит лишь его незначительное количество, поэтому обычно непосредственно под костью визуализируется большая область чёрного цвета, кроме которой больше ничего не видно. Это явление называется "акустическая тень". [18]
На современном оборудовании для ультразвуковых исследований, чтобы визуализировать на снимке область чёрного цвета, находящуюся непосредственно под костью, определяют сильную отражающую волну, после чего автоматически усиливают возвращающийся ультразвуковой сигнал и пытаются визуализировать структуру, которая скрывается в акустической тени. [2] На ультразвуковом снимке длинных трубчатых костей при гипофосфатазии с обеих сторон диафиза в нижней части видны дорожки. Предполагается, что это происходит из-за того, что усиливается ультразвуковой сигнал от метафиза, через который проходит относительно большое количество ультразвуковых лучей. [21]
Диагностика детской и взрослой гипофосфатазии
В более старшем возрасте диагноз можно заподозрить по клиническим проявлениям.
При подозрении на гипофосфатазию определяется уровень щелочной фосфатазы в крови. Приняты следующие нормы щелочной фосфатазы:
УЗИ, рентгенограмма при гипофасфатазии у плода
Гипофосфатазия (ГФФ) - редкое наследственное жизнеугрожающее заболевание, вызванное недостаточностью фермента тканенеспецифической щелочной фосфатазы (ТНЩФ), кодируемой геном ALPL, сопровождающееся характерной клинической картиной и низким уровнем активности щелочной фосфатазы в сыворотке крови. Частота встречаемости ГФФ варьирует в различных странах, и для российской популяции оценивается как 1:100 000. Наиболее частыми клиническими проявлениями ГФФ в детском возрасте являются задержка роста, нарушение минерализации костей, рахитоподобные деформации скелета, низкий мышечный тонус, утомляемость при физической нагрузке, болевой синдром в виде миалгии и оссалгии, повторные переломы костей, выпадение зубов, дыхательные расстройства, пиридоксин-зависимые судороги, гиперкальциемия, нефрокальциноз. Полиорганный характер поражения и выраженные метаболические расстройства приводят в некоторых случаях к тяжелым жизнеугрожающим состояниям. Смертность при ГФФ с ранним дебютом составляет от 50 до 100%.
В настоящей работе представлен результат ретроспективного исследования для идентификации пациентов с редким заболеванием ГФФ. Для исследования был разработан диагностический алгоритм, учитывающий современные референтные значения активности ТНЩФ для детей, скрининг базы тестов и анализ клинических симптомов. В результате проведенной работы обнаружены 3 пациента с ГФФ, у которых заболевание подтверждено генетически.
Приведенный алгоритм может быть использован для эффективной диагностики ГФФ в других лечебных учреждениях без дополнительных финансовых затрат.
Ключевые слова: гипофосфатазия, ГФФ, тканенеспецифическая щелочная фосфатаза, рахитоподобные состояния, скелет, детский возраст, диагностика, скелетные деформации, низкорослость, генетическая диагностика, ALPL.
Retrospective diagnostics of hypophosphatasia in children
Sosnina I.B. 1 , Sukcheev M.B., Ivashikina T.M. 1 , Snegova E.V. 1 , Pashkova V.P. 1 , Klimenkova O.A. 1 , Pervunina T.M. 2
1 Consultative Diagnostic Center for Children, St. Petersburg
2 V.A. Almazov Federal North-West Medical Research Center, St. Petersburg
Hypophosphatasia is a rare inherited life-threatening condition resulting from the deficiency of tissue-specific alkaline phosphatase (AP) encoded by APL gene. This disorder with specific clinical manifestations is characterized by low serum activity of AP. The occurrence of hypophosphatasia varies among the countries, its estimated rate in Russia is 1 per 100,000 population. The most common symptoms of hypophosphatasia in children are defective bone mineralization, rachitic skeletal deformations, low muscle tonus, fatigue after physical activity, pains (myalgia, ossalgia), recurrent bone fractures, losing teeth, respiratory disorders, pyridoxine-dependent seizures, hypocalcaemia, and nephrocalcinosis. Multiple organ dysfunction and severe metabolic disorders lead to life-threatening conditions. Early hypophosphatasia mortality is 50-100%.
The publication addresses the outcomes of retrospective study on the identification of patients with this rare disease. Diagnostic algorithm considers current reference values of tissue-nonspecific AP activity in children and includes test database screening and clinical symptom assessment. As a result, three patients with genetically verified hypophosphatasia were identified. This algorithm can be used for precise cost-effective diagnosis of hypophosphatasia in other clinics.
Key words: hypophosphatasia, HPP, tissue-nonspecific alkaline phosphatase, rickets like conditions, skeleton, children, diagnostics, skeletal deformations, stunting, genetic diagnostics, ALPL.
For citation: Sosnina I.B., Sukcheev M.B., Ivashikina T.M. et al. Retrospective diagnostics of hypophosphatasia in children // RMJ. 2016. № 26. P. 1778-1781.
В статье представлен опыт ретроспективной диагностики гипофосфатазии у детей
Введение
Тканенеспецифическая щелочная фосфатаза (ТНЩФ) - это фермент из группы гликопротеинов, катализирующих гидролиз фосфодиэфирной связи с высвобождением неорганического фосфата [1]. ТНЩФ экспрессируется на поверхности клеток и представлена в различных органах и тканях человека, включая костную ткань, печень и почки. Сниженная активность ТНЩФ приводит к широкому спектру клинических проявлений.
ГФФ - это редкое наследственное жизнеугрожающее заболевание, при котором активность ТНЩФ снижена. ГФФ вызвана нарушением функции гена ALPL, кодирующего ТНЩФ, что приводит к ее недостаточности [2]. В норме ТНЩФ регулирует обмен неорганического пирофосфата (НПФ) и поступление витамина B6 в нервные клетки [1].
При ГФФ НПФ не разрушается, его концентрация увеличивается, и прекращается рост кристаллов гидроксиапатита 4, что вызывает нарушения в формировании костной ткани и скелетные деформации [1, 6]. В то же время НПФ в повышенной концентрации образует связи с ионами кальция с формированием кристаллов пирофосфата кальция, который может накапливаться в почках (вызывает нефрокальциноз) или суставах (вызывает боли в суставах и развитие кристаллического артрита и псевдоподагры) [7].
Важной функцией ТНЩФ в работе нервной системе является регулирование поступления витамина В6 в ткани головного мозга. ТНЩФ отщепляет фосфат от пиридоксаль-5-фосфата (П5Ф, форма витамина B6) с образованием пиридоксаля, который способен проникать через клеточные мембраны в ЦНС, где происходит повторное присоединение фосфата к пиридоксалю с образованием П5Ф. П5Ф выступает кофактором нейротрансмиттеров (серотонина, допамина, гамма-аминомасляной кислоты и др.), а его дефицит в ЦНС при ГФФ выражается в виде судорог, которые купируются введением витамина B6 [8, 9]. Наличие судорог при ГФФ служит прогностическим признаком высокой вероятности летального исхода [10]. Кроме того, аномалии формирования костей грудной клетки становятся одной из причин гипоплазии легких, что может приводить к дыхательной недостаточности у пациентов с ГФФ [11].
Тяжелые формы ГФФ чаще всего вызваны наличием гомозиготной или компаунд-гетерозиготной мутации в гене ALPL (OMIM 171760) [12]. ТНЩФ экспрессируется на поверхности клеток как гомодимер, поэтому некоторые гетерогизогные мутации могут приводить к доминантно-негативному эффекту со снижением активности гомодимерной молекулы фермента [13, 14]. Мутация даже в одном аллеле может приводить к развитию ГФФ [15]. Носители одинаковой мутации в семье могут иметь различные степени проявления ГФФ, что указывает на наличие модулирующих факторов, участвующих в метаболизме фосфатов. В некоторых случаях ГФФ мутации в гене ALPL не удается обнаружить стандартными методами генетической диагностики [16], поэтому для постановки диагноза ведущими критериями являются клинические признаки ГФФ и сниженная активность ТНЩФ (ниже нижней границы нормы для данного возраста и пола) 20.
Клинические формы ГФФ
В зависимости от возраста, в котором появляются первые симптомы, ГФФ условно разделяют на несколько форм 23. Перинатальная форма (иногда называемая перинатальной летальной формой ГФФ) диагностируется при беременности на УЗИ или сразу после рождения. При инфантильной форме ГФФ первые симптомы заболевания обнаруживаются в первые 6 мес. жизни. Для таких пациентов характерна дыхательная недостаточность, судороги, обусловленные дефицитом витамина В6, скелетные деформации, переломы (иногда полученные внутриутробно) и отставание в развитии [21]. При биохимическом обследовании у пациентов с перинатальной и инфантильной формами ГФФ обнаруживается гиперкальциемия, которая может приводить к развитию нефрокальциноза [22]. Неправильное формирование костей черепа приводит к краниосиностозу и повышению внутричерепного давления, что может потребовать нейрохирургического лечения [16].
Если признаки ГФФ проявились после 6 мес. жизни, такую форму заболевания принято называть детской формой. Для нее наиболее характерны задержка роста и моторного развития, дефицит массы тела, рахитоподобные изменения, скелетные деформации, ранняя потеря зубов с интактными корнями (в среднем до 5 лет). Кроме того, пациенты с детской формой ГФФ подвержены риску частых переломов костей [21, 22].
Для взрослой формы ГФФ характерен широкий спектр проявлений: остеопорозные изменения скелета, «утиная походка», боли в мышцах, частые плохо заживающие переломы, отложения кристаллов в суставах [21, 22, 24, 25].
Единственным клиническим проявлением одонтогипофосфатазии является раннее выпадение зубов с интактным корнем [21, 22, 26].
Данные о распространенности заболевания различаются в разных странах, но предполагаемая средняя распространенность тяжелых форм ГФФ примерно 3,3 случая на 1 млн новорожденных. Рассчитанный уровень заболеваемости средними формами ГФФ 1:6000 [12]. В России ожидаемая распространенность 1:100 000 [27].
Ретроспективная диагностика ГФФ
В настоящее время известно около 7000 редких заболеваний, которые поражают примерно 5% человеческой популяции [28, 29]. По статистике, пациент с редким заболеванием получает правильный диагноз в среднем через 5 лет после наступления первых симптомов [29] и после консультаций примерно у 7 врачей разных специальностей [30]. В то же время около 30% пациентов с редкими заболеваниями умирают, не достигнув 5-летнего возраста. Поэтому своевременная диагностика редких заболеваний критически необходима для начала адекватного ведения пациентов. При ГФФ с ранним наступлением первых симптомов 73% умирают в течение первых 5 лет жизни [28], а пациенты с детской формой ГФФ подвержены риску инвалидизации в последующие годы [22, 31, 32]. Кроме того, терапия ГФФ при неправильной диагностике может приводить к дополнительному ухудшению состояния пациентов [33, 34]. Разнообразие клинических проявлений ГФФ, а также распространенный во врачебной практике недостаток внимания к пониженной активности ТНЩФ и отсутствие установленных педиатрических норм активности ТНЩФ ведут к значительной гиподиагностике ГФФ в РФ [20].
Чтобы провести ретроспективную диагностику ГФФ среди пациентов детского возраста, в Консультативно-диагностическом центре для детей (КДЦД) (Санкт-Петербург) были установлены педиатрические референтные значения активности ТНЩФ, которые значительно зависят от возраста. Значения активности ТНЩФ взяты из международного исследования CALIPER [32], в котором были установлены референтные значения нескольких десятков биохимических маркеров в зависимости от возраста, а также из клинических рекомендаций по диагностике ГФФ, разработанных в РФ (табл. 1) [20].
Алгоритм ретроспективной диагностики
В КДЦД проведен ретроспективный анализ лабораторных данных пациентов в возрасте от 0 до 18 лет, обратившихся к специалистам центра в 2015 г. (около 560 тыс. тестов) (рис. 1). Было отобрано для исследования 280 амбулаторных карт пациентов, имеющих показатели ТНЩФ ниже установленных возрастных значений. С целью определения необходимости дальнейшего генетического тестирования было проведено сопоставление жалоб, клинической картины и лабораторных данных пациентов с клиническими критериями гипофосфатазии. В результате выборки выявлено 9 случаев с наличием в клинической картине характерных для гипофосфатазии симптомов в виде задержки роста, костных деформаций, болей в ногах. Все 9 пациентов были приглашены на осмотр для оценки клинической картины и решения вопроса о целесообразности проведения исследования на наличие мутации в гене ALPL. Из приглашенных явились на осмотр 6 пациентов. Пациенты и их родители, проинформированные о цели исследования, выразили добровольные согласие на участие в исследовании. Произведен забор венозной крови в пробирку с ЭДТА для направления на генетическую диагностику. У 2-х пациентов выявлены патогенные мутации в гене ALPL и поставлен диагноз «гипофосфатазия». Диагноз подтвержден повторными независимыми исследованиями в лабораториях, осуществивших также генетическое тестирование родителей пациентов.
Предварительный анализ тестов с низкой активностью ТНЩФ показал, что примерно в 2/3 случаев это были пациенты гастроэнтерологического профиля с различными заболеваниями ЖКТ (целиакия, лактазная недостаточность и другие ферментопатии, реконвалесценты перенесенных кишечных инфекций) и примерно в 1/3 случаев - пациенты эндокринологического профиля (в основном с диагнозом «гипотиреоз»).
Установка педиатрических референтных значений ТНЩФ позволила диагностировать также одного пациента с ГФФ проспективно в течение 2016 г. Диагноз ГФФ подтвержден при помощи генетического исследования гена ALPL, где была найдена патогенная мутация (c. 571G>A, p. E191K), ранее описанная в литературе как патогенная (см. рис. 1) [33].
Описание клинических случаев ГФФ Клинический случай № 1
Пациент Г.П., 2009 г. р., обратился с жалобами на задержку роста с 5 лет. Анамнез жизни: роды 2-е, на 36-й неделе (5-я беременность, токсикоз 2-й половины, гестоз, резус-отрицательная мать, носитель HCV), вес при рождении - 2850 г, рост - 51 см, оценка по шкале Апгар - 7/8 баллов. При рождении установлен диагноз: внутриутробная гипотрофия, перинатальная энцефалопатия, внутриутробная инфекция. В семейном анамнезе обнаружен низкий рост у бабушек по линии отца и матери (150 см), рост отца - 171 см, матери - 161 см. Пациент проконсультирован эндокринологом в 6 лет. Данные пациента при осмотре: рост - 105 см (отставание на 7 см), вес - 14,8 кг (дефицит массы тела - 13,4%). По данным рентгенограммы кистей рук: развитие скелета соответствует 3-м годам, турецкое седло - без патологии. УЗИ органов брюшной полости: уплотнение стенок желчного пузыря. УЗИ щитовидной железы: без патологии, объем железы - 2,3 см3 (соответствует норме). Эндокринологом диагностированы первичный субклинический идиопатический гипотиреоз и низкорослость. Обследование по гастроэнтерологическому профилю позволило исключить паразитозы, хроническую кишечную инфекцию. В гемограмме обнаружен сниженный уровень половых гормонов.
Тестирование крови на гипофосфатазию выявило снижение уровня щелочной фосфатазы до 88 Ед/л (возрастная норма 142-335 Ед/л). В результате генетического обследования пациента выявлен дефект гена ALPL, аутосомно-доминантный тип наследования (мутация с. 571G>A в гетерозиготном состоянии) [33]. Результаты генетического обследования родителей: у матери мутация с. 571G>A в гетерозиготном состоянии в гене ALPL; у отца мутация с. 571G>A в гене ALPL отсутствует. В качестве дополнительного исследования проведено измерение концентрации субстрата ТНЩФ П5Ф в цельной крови пациента: 44 мкг/л (возрастная норма - 8,7-27,2 мкг/л).
Клинический случай № 2
Пациент И.Ш., 2008 г.р. Задержка роста и отставание в физическом развитии отмечались с раннего возраста. Анамнез жизни: родился от 1-й беременности на сроке 35 нед. Вес при рождении - 2400 г, рост - 46 см. Синдром дыхательных расстройств, дыхательная недостаточность II-III степени, внутриутробная инфекция с поражением легких. Проводилась дыхательная поддержка с помощью искусственной вентиляции легких. В раннем возрасте отмечались частые ОРВИ, отиты, бронхиты, а также ветряная оспа, пищевая аллергия, атопический дерматит и аллергический ринит. Семейный анамнез: рост матери - 158 см, вес - 40 кг, рост отца - 168 см, ожирение II степени. Данные осмотра пациента в 8 лет: рост -114 см, вес - 18,2 кг. По данным рентгенографического исследования кисти: развитие скелета соответствует 5-6 годам, турецкое седло без патологии. Исследование водно-солевого баланса: без патологии. Инсулиноподобный фактор роста - 128,154 мкм/л, кортизол - 272,8 нмоль/л, фосфор - 1,34 ммоль/л, кальций - 4,9 ммоль/л, ионизированный кальций - 1,32 ммоль/л. Уровень активности ТНЩФ в сыворотке крови - 38 Ед/л (возрастная норма - 142-335 Ед/л). Диагноз «гипофосфатазия» подтвержден в результате генетического исследования: выявлена гетерозиготная мутация с. 436G>A, р. Е146К (rs138587317, частота минорного аллеля 0,02%) [33].
Заключение
Приведенный в данной публикации алгоритм ретроспективной диагностики редкого заболевания ГФФ позволил без дорогостоящего массового скринига и привлечения дополнительных ресурсов применить ранее полученные биохимические и клинические данные и выявить группу пациентов с высокой вероятностью положительного результата.
В итоге выявлены 2 пациента с низкорослостью и рахитоподобными изменениями скелета, у которых диагноз «гипофосфатазия» подтвержден генетически. Кроме того, после установления педиатрических норм активности ТНЩФ обнаружен еще один пациент с низким уровнем активности ТНЩФ, клиническая картина у которого соответствовала ГФФ. Секвенирование гена, кодирующего ТНЩФ, показало наличие описанной ранее патогенной мутации. В последующем данным пациентам может быть оказана медицинская помощь соответственно диагнозу.
Таким образом, применение теста на активность щелочной фосфатазы, входящего в различные панели и часто назначаемого для диагностики, а также алгоритма клинического анализа позволяют обнаружить пациентов с редким заболеванием ГФФ. Адаптация данного алгоритма ретроспективной диагностики в других ЛПУ может способствовать улучшению качества диагностики и медицинской помощи без применения дополнительных средств.
ФГБУ «Эндокринологический научный центр», Москва
Эндокринологический научный центр, Москва
ГБОУ ВПО «Северный государственный медицинский университет», Архангельск, Россия
Гипофосфатазия: клиническое описание трех случаев заболевания с молекулярно-генетической верификацией диагноза
Журнал: Проблемы эндокринологии. 2015;61(3): 37‑42
Гипофосфатазия — редкое наследственное рахитоподобное заболевание, обусловленное снижением активности тканенеспецифической щелочной фосфатазы, кодируемой геном ALPL. Различают несколько форм данного заболевания в зависимости от тяжести течения и возраста манифестации. Основными клиническими признаками гипофосфатазии являются рахитические деформации костей скелета, мышечная гипотония и дыхательная недостаточность в раннем детском возрасте, задержка физического и моторного развития, раннее выпадение зубов. В зрелом возрасте наблюдаются стресс-переломы, мышечные боли, кальцификация связок и суставов. Биохимическими маркерами патологии служат сниженный уровень щелочной фосфатазы, повышенное содержание фосфоэтаноламина в моче; при тяжелых формах заболевания — гиперкальциемия, гиперфосфатемия, снижения уровня паратгормона. Приведено клиническое описание пациентов с гипофосфатазией различной тяжести, у которых впервые в России проведена молекулярно-генетическая верификация диагноза.
Гипофосфатазия (ГФ, OMIM 146300, 241500, 241510) — редкое наследственное нарушение метаболизма, в основе которого лежит снижение активности фермента тканенеспецифической щелочной фосфатазы (ЩФ), кодируемой геном ALPL. Клиническая картина и степень тяжести ГФ варьируют в широких пределах; четкой корреляции между генотипом и фенотипом не выявлено. В зависимости от возраста манифестации заболевания и тяжести симптомов различают шесть клинических форм: перинатальную, доброкачественную пренатальную, инфантильную, детскую, взрослую, oдонтогипофосфатазию (табл. 1).

Таблица 1. Клинические формы гипофосфатазии
Диагноз ГФ впервые был установлен D. Rathbun [1] на основании выраженных рахитических изменений костей у ребенка и парадоксально низкого уровня ЩФ.
Распространенность тяжелых форм ГФ (перинатальная и инфантильная) оценивается в 1 на 100 000 новорожденных (в основном англосаксонского происхождения); эти формы наиболее распространены в изолированной популяции меннонитов Канады, где частота встречаемости тяжелых форм составляет 1 на 2500 новорожденных, что, возможно, объясняется «эффектом основателя гена» [2, 3]. Более мягкий вариант заболевания (взрослая форма) встречается с частотой примерно 1: 6000 человек [3]. В России распространенность данного заболевания неизвестна.
Ранее в отечественной литературе [4] сообщалось о пренатальной диагностике ГФ на сроке 37 нед гестации у плода с выраженной деминерализацией скелета. Нами впервые в России проведено молекулярно-генетическое подтверждение диагноза у пациентов с ГФ различной тяжести, у 3 детей выявлены составные гетерозиготные мутации гена ALPL.
Материал и методы
Проведена оценка анамнеза, клинических данных пациентов, анализ лабораторных и рентгенографических показателей.
Результаты
Пациент 1. Г. М., мальчик, 4 года, рожден на 39—40-й неделе гестации с нормальным ростом и массой тела (53 см, 3800 г) от здоровых родителей, состоящих в неродственном браке. С рождения отмечалась выраженная гипотония, в результате чего ребенок отставал в моторном развитии: поздно начал держать голову и сидеть. Ходить «утиной» походкой мальчик начал с 1 года 7 мес; отмечалась быстрая утомляемость и боли в ногах при ходьбе. В возрасте 2 лет у ребенка выпали передние молочные зубы. В связи с отставанием в росте в 3 года с целью исключения cоматотропной недостаточности проведена СТГ-стимуляционная проба с клофелином; результат нормальный (максимальный выброс СТГ 16,1 нг/мл).
При обследовании в ФГБУ «Эндокринологический научный центр» (ЭНЦ) в возрасте 4 лет отмечалось отставание в физическом развитии: рост 91,3 см (SDSр.=-2,66), масса тела 11,3 кг (SDS ИМТ =-2,1) (рис. 1). Обращали на себя внимание скафоидная форма черепа за счет краниостеноза, плосковальгусная установка стоп, «переваливающаяся» походка, быстрая утомляемость, в связи с чем ребенок чаще находился в коляске. При биохимическом анализе крови был двукратно обнаружен низкий уровень ЩФ — до 62,8 Ед/л (норма 180—720 Ед/л), на фоне нормальных показателей кальция, витамина В 6 , умеренном повышении содержания фосфора и незначительно сниженном уровне паратгормона (табл. 2). При рентгенологическом исследовании выявлено значительное разряжение костной ткани в области метафизов локтевой и лучевой костей (рис. 2).
Таблица 2. Биохимические параметры сыворотки крови пациентов
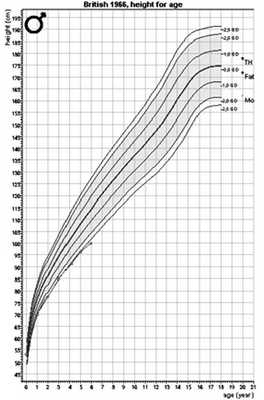
Рис. 1. График роста пациента 1.
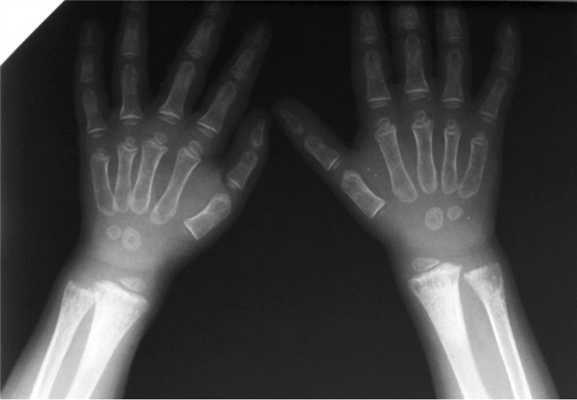
Рис. 2. Рентгенограмма костей верхних конечностей пациента 1. Выраженное разряжение зон метафизов лучевых и локтевых костей.
В качестве наиболее вероятной причины раннего выпадения молочных зубов, нарушения походки, низкого уровня ЩФ крови, рахитических изменений структуры костей был рассмотрен диагноз Г.Ф. Данный диагноз был подтвержден результатами молекулярно-генетического анализа гена ALPL. Выявлено наличие 2 гетерозиготных мутаций: c.340G>A p. A114T (замена аланина на треонин в позиции 114), и c.571G>A p. E191K (замена глутаминовой кислоты на лизин в позиции 191). Выявленные мутации были описаны ранее [1, 5—7].
Пациент 2. В.М., девочка, 8 лет. Данный клинический случай постановки диагноза «ГФ, инфантильная форма» ребенку 2 года 8 мес на основании клинической картины был описан ранее [8]. При обследовании в ЭНЦ у девочки 8 лет обнаружены дефицит массы тела: масса тела 17,2 кг (SDS ИМТ =-3,24), рост 117,1 см (SDSр.=-1,75) (рис. 3), долихоцефалическая форма черепа, выраженная деформация грудной клетки, рахитические «браслетки» на руках и варусная деформация бедренных костей. Содержание Щ.Ф. в крови было значительно сниженным (до 21 Ед/л), уровень фосфора умеренно повышен (см. табл. 2). При рентгенологическом исследовании выявлены характерные рахитические изменения структуры костей верхних и нижних конечностей: неровность метафизов, узурация эпифизов. Молекулярно-генетический анализ выявил наличие составной гетерозиготной мутации гена ALPL: c.253A>C p. T85P с заменой треонина на пролин в позиции 85 и c.571G>A p. E191K. Вторая мутация оказалась аналогичной таковой у первого пациента.
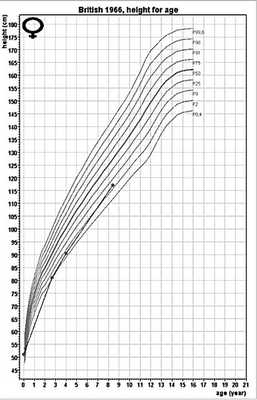
Рис. 3. График роста пациента 2.
Пациент 3. В.М., девочка, 3 года. На 24-й неделе гестации при УЗИ были выявлены признаки скелетной дисплазии трубчатых костей. Ребенок родился в срок с нормальными росто-весовыми показателями (масса тела 3465 г, длина тела 52 см), без признаков нарушения дыхания и деформаций скелета. На первом году жизни отмечалось выраженное отставание в росте (в 1 год рост 66 см, SDSр.=-3,0) (рис. 4), задержка моторного развития ребенка — начала сидеть с 9 мес, ходить с 18 мес. Первые зубы прорезались в 9 мес, однако с 14 мес выпали два нижних резца, а в 2 года 8 мес — верхние резцы. С 1,5 лет наблюдались частые респираторно-вирусные заболевания с пневмонией. При обследовании в возрасте 3 лет: масса тела 10,5 кг (SDS ИМТ =-2,31), рост 88 см (SDS=-1,31), вальгусная деформация нижних конечностей. В крови низкий уровень ЩФ (до 40 Ед/л). При рентгенологическом исследовании определялись множественные разнокалиберные участки разрежения костной ткани по типу «матового стекла» в зонах метафизов костей предплечий и костей голеней, а также булавовидные расширения передних отрезков ребер. Молекулярно-генетический анализ показал наличие составной гетерозиготной мутации гена ALPL: c.571G>A p. E191K (аналогичной таковой в двух первых случаях), и с.1166С>Т р. Т389I (замена треонина на изолейцин в позиции 389).
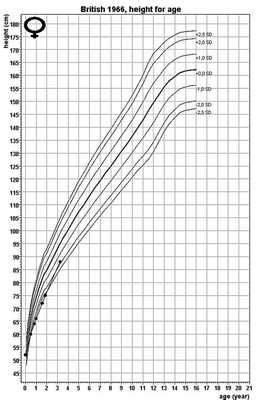
Рис. 4. График роста пациента 3.
Термином ЩФ обозначают группу ферментов, расположенных на мембране клеток и принимающих участие в расщеплении эфиров фосфорной кислоты (фосфогидролазы). Источниками различных изоформ ЩФ являются костная ткань, печень, почки, плацента (ALPP), слизистая оболочка тонкого кишечника (ALPI), эмбриональная ткань. Ген ALPL (OMIM 171760) кодирует тканенеспецифическую ЩФ (TNSALP), экспрессирующуюся в печени, костях и почках [9]. Ген расположен на коротком плече хромосомы 1 (1p36.1—р.34) и состоит из 12 экзонов. Основная функция TNSALP сводится к гидролизу монофосфатных эфиров, включая неорганические пирофосфаты (PPI), фосфоэтаноламин (РЕА), пиридоксаль-5-фосфат, которые являются физиологическими ингибиторами образования кристаллов гидроксиапатита костного матрикса. Клиническая картина заболевания обусловлена нарушением минерализации костной ткани вследствие накопления пирофосфатов в крови из-за снижения ферментативной активности TNSALP. Перинатальная форма ГФ характеризуется практически полным отсутствием минерализации костей скелета, наличием остеохондральных шпор, выступающих через кожу предплечий и ног, значительным укорочением длинных трубчатых костей, судорожным синдромом (витамин В6-зависимые судороги). Данная форма имеет высокий риск летальности внутриутробно или в первые месяцы жизни из-за выраженной дыхательной недостаточности вследствие гипоплазии легких и биомеханических нарушений акта дыхания из-за рахитических изменений грудной клетки. В редких случаях у пациентов с доброкачественной пренатальной формой происходит спонтанное улучшение минерализации.
Инфантильная форма ГФ манифестирует с первого полугодия жизни рахитическими деформациями грудной клетки и выраженной мышечной гипотонией, которые обусловливают нарушение дыхания, частые воспалительные процессы в легких, задержку моторного развития. Гиперкальциемия является причиной плохой прибавки в массе тела, рвоты, склонности к запорам, жажды, полиурии, раннего закрытия большого родничка и швов черепа, что приводит к краниостенозу, повышению внутричерепного давления. Гиперкальциурия способствует развитию нефрокальциноза. Летальность при данной форме составляет до 50% к первому году жизни.
Детская и взрослая формы имеют более благоприятное течение и характеризуются рахитическими изменениями скелета, низким ростом, болью в мышцах и связках, краниостенозом, формированием долихоцефалической формы черепа, ранним выпадением молочных (до 5 лет) и постоянных зубов, изменением походки («переваливающаяся» походка), хондропатиями или псевдопереломами в результате остеопении и отложения пирофосфатата кальция в связках и суставах в зрелом возрасте. Биохимическими маркерами патологии служат умеренно или резко сниженный уровень щелочной фосфатазы, повышение концентрации пиридоксаль-5-фосфата в крови и фосфоэтаноламина в моче; в раннем детском возрасте можно обнаружить гиперкальциемию, гиперфосфатемию, снижение уровня паратгормона.
Для пациентов с наиболее легкой формой (одонтогипофосфатазия) характерно только раннее выпадение зубов или развитие частого кариеса из-за увеличения камер пульпы или корневых каналов, однако биохимические маркеры заболевания остаются такими же.
К настоящему времени описано более 280 мутаций гена ALPL [10], большинство из которых составляют миссенс-мутации (до 80%), реже небольшие делеции гена (10,5%), нарушения сплайсинга (6%) и нонсенс-мутации (4%). Наиболее тяжелые формы наследуются аутосомно-рецессивным путем, при более легких формах может иметь место и доминантный тип наследования [6, 7, 11]. У трансгенных мышей с гомозиготной мутацией гена ALPL активность ЩФ практически полностью отсутствовала (менее 1%), а у гетерозигот снижена на 50% [12].
Интересно, что выявленная мутация p. A114T гена ALPL у первого пациента оказалась аналогично той, что была обнаружена в гетерозиготном состоянии у матери мальчика, которому впервые в мире был установлен диагноз ГФ [1]. Поскольку у матери имелась миссенс-мутация p. A114T гена ALPL, а у отца — миссенс-мутации p. D294A в 9 экзоне и полиморфизм в 12 экзоне (Т1565С; Val505Ala), было высказано предположение, что ребенок имел компаунд-гетерозиготные мутации гена, обусловившие летальную форму патологии; при этом у родителей клинические признаки заболевания отсутствовали.
Можно предположить, что у второго нашего пациента раннее начало заболевания и более тяжелое течение обусловлены более выраженным снижением активности ЩФ в результате мутаций p. T85P/р.E191K, чем у мальчика с дефектом p. A114T/p.E191K.
Выявленная нами мутация р. Т389I в экзоне 10 гена ALPL была ранее описана у ребенка с перинатальной формой ГФ, который имел компаунд-гетерозиготные мутации р. Т389I/р.S368del (Versailles lab oct., 2004). Клиническая картина заболевания у нашего пациента с дефектом p. E191K/р.Т389I гена ALPL больше соответствует инфантильной форме патологии.
Разнообразие клинических признаков и форм ГФ требует рассмотрения данного диагноза у пациентов среднего возраста с жалобами на частые и плохо срастающиеся переломы, скелетно-мышечные боли. Как правило, у пациентов с взрослой формой ГФ в анамнезе имеется указание на проявления рахита в детстве или раннее выпадение зубов (молочных или постоянных). Переломы обычно локализуются в лучевой, плечевой костях; у женщин — подвертельные переломы бедренных костей, стресс-переломы плюсневых костей, у мужчин — переломы позвоночника [14]. Отложение пирофосфата кальция в связках, сухожилиях и суставах может быть причиной болевого синдрома и ограничения двигательной активности [15]. Описан семейный случай ГФ, единственным проявлением заболевания у пациентов был болевой синдром на фоне кальцификации связок и суставов [15]. Правильная диагностика может предотвратить неоправданное использование бисфосфонатов, которые применяются при остеопорозе. При Г.Ф. данные препараты утяжеляют заболевание, так как являются аналогами пирофосфатов, избыточно накапливающихся при снижении активности ЩФ.
В 1956 г. для лечения ГФ с ограниченным успехом применяли кортизон. Использование заменного переливания плазмы богатой ЩФ от пациентов с болезнью Педжета было предпринято в 80-х годах XX века. Пересадка аллогенных мезенхимальных стволовых клеток донора, изолированная трансплантация остеобластов также не давали достаточного эффекта [16]. У взрослых пациентов применение терипаратида (рекомбинантного паратгормона) было малоэффективно. В настоящее время имеются данные об использовании заместительной ферментной терапии рекомбинантной тканенеспецифической Щ.Ф. Эффективность такого лечения доказана на животных моделях и в клинических испытаниях на людях [17, 18]. Применение данного препарата у 12 пациентов с перинатальной и инфантильной формами ГФ значительно улучшило структуру костей, что оказало положительное влияние на функцию легких и двигательную активность детей.
В заключение необходимо подчеркнуть важность дифференциальной диагностики различных рахитоподобных заболеваний с Г.Ф. Решающее значение в постановке диагноза ГФ имеет правильная оценка клинических и анамнестических данных, определение уровня ЩФ с учетом возрастных норм и молекулярно-генетический анализ.
Читайте также:
- Искусственный климат в космосе. Физиологические проблемы невесомости
- Плазмоцитома сосудистой оболочки глаза: признаки, гистология, лечение, прогноз
- Грипп: симптомы, признаки, диагностика и лечение
- Тонкоигольная пункционная аспирационная биопсия щитовидной железы. Характеристика образований щитовидной железы
- Синдром Вагнера-Унферрихта (Wagner-Unverricht)