Ведение больных с периодическим параличом. Профилактика периодического паралича.
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
У большинства больных с периодическим параличом, у которых во время приступов заболевания содержание калия в сыворотке крови остается на нормальном уровне, проявляются такие же симптомы, как и при гиперкалиемическом периодическом параличе; они также хорошо реагируют на препараты калия, фактически так называемые гиперкалиемические и нормокалиемические формы периодического паралича по сути являются одной и той же нозологической единицей. Изредка больные с нормокалиемическим периодическим параличом оказываются нечувствительными к препаратам калия, однако при мышечной биопсии у них обнаруживают признаки деструкции мышцы и другие морфологические изменения, что позволяет исключить первичный периодический паралич.
Что провоцирует / Причины Нормокалиемического паралича:
Наследуется по аутосомно-доминантному типу. Провоцирующими факторами являются продолжительный сон, длительное пребывание в одной позе, переохлаждение.
Симптомы Нормокалиемического паралича:
Болезнь проявляется до 10-летнего возраста. Особенностью ее является сравнительно медленно (в течение нескольких суток) пароксизмально нарастающая умеренная слабость в мышцах туловища, конечностей и в жевательной мускулатуре, а также медленный (1-2 нед) регресс симптоматики.
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют.
Диагностика Нормокалиемического паралича:
Диагноз строится на основании генеалогического анализа, особенностей клинической картины, с учетом возраста, в котором начинается заболевание, времени возникновения пароксизма (ночью, утром, днем, в неопределенное время), степени выраженности мышечной слабости, частоты и длительности приступа, провоцирующих факторов, данных лабораторного биохимического исследования (содержание биоэлектрической активности мышц).
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний, - тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Лечение Нормокалиемического паралича:
Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
К каким докторам следует обращаться если у Вас Нормокалиемический паралич:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Нормокалиемического паралича, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Пароксизмальная миоплегия
Пароксизмальная миоплегия (периодический паралич) — нервно-мышечное заболевание, характеризующееся периодически возникающими пароксизмами преходящего паралича скелетной мускулатуры, обуславливающего обездвиженность. Наблюдается в трех вариантах: гипо-, гипер- и нормокалиемическая форма. Диагностика миоплегии включает неврологический осмотр, генетическое консультирование, биохимию крови, электромиографию, при необходимости — биопсию мышц и провокационную пробу с хлоридом калия. Лечебная тактика в период миоплегического пароксизма зависит от формы периодического паралича. В период между приступами необходимо соблюдение диеты, избегание провоцирующих факторов и прием диуретиков (спиронолактона, ацетазоламида, гидрохлоротиазида).
Общие сведения
Патогенез пароксизмальной миоплегии
В настоящий период как механизм развития миоплегических приступов, так и патогенез всего заболевания в целом остается предметом изучения медиков, биохимиков и физиологов во всем мире. Ранее существовала гипотеза Конна, согласно которой основу заболевания составляет периодически возникающая повышенная экскреция альдостерона — перемежающийся гиперальдостеронизм. Ее подтверждением считали положительный эффект от терапии антагонистами альдостерона. Однако недавно было доказано, что сдвиги в концентрации альдостерона, которыми сопровождается пароксизмальная миоплегия, имеют вторичный характер.
Имеются предположения о генетически детерминированном нарушении проницаемости мембран миофибрилл и клеточных оболочек других структурных элементов в поперечно-полосатой мышечной ткани, в результате которого ионы натрия и вода проходят в миофибриллы, где скапливаются в вакуоли. Изменение содержания натрия в сыворотке крови приводит к соответствующему изменению концентрации калия. Следствием этого дисбаланса является нарушение внутриклеточных обменных процессов (в т. ч. обмена гликогена) в мышечной ткани и мионевральных синапсах, происходит блокировка нервно-мышечной передачи, миофибриллы теряют способность к сокращению. При возвращении ионного равновесия в норму функция миофибрилл и нервно-мышечных синапсов восстанавливается и мышцы вновь приобретают способность сокращаться.
Гипокалиемическая пароксизмальная миоплегия
Встречается чаще других. Является аутосомно-доминантно наследуемым заболеванием с неполной пенетрантностью гена. Однако нередки спорадические случаи миоплегии. Пик заболеваемости приходится на возрастную группу 10-18 лет, хотя случаи периодического паралича наблюдались и у лиц старше 30 лет. У мужчин отмечается чаще, чем у женщин. Вне пароксизма ни субъективные, ни объективные признаки заболевания не определяются. Спровоцировать приступ может переедание, употребление чрезмерного количества углеводов или поваренной соли, физическое перенапряжение, прием алкоголя. У женщин пароксизмальная миоплегия обычно связана с менструальным циклом — приступы зачастую наблюдается в 1-й день менструации или за 1-2 дня до ее начала.
Миоплегический пароксизм обычно начинается в утреннее или ночное время. Проснувшись пациенты обнаруживают себя парализованными. Миоплегия распространяется на скелетные мышцы всего тела, конечностей и шеи, при тяжелом течении — на дыхательную и лицевую мускулатуру. Типична резкая мышечная гипотония, выпадение всех сухожильных рефлексов. Приступ сопровождается вегетативной симптоматикой — тахикардией, гиперемией лица, гипергидрозом, полидипсией, учащенным дыханием. Его длительность варьирует от часа до 1-2 суток, но чаще составляет 2-4 часа. К концу пароксизма отмечается постепенное нарастание мышечной силы, начинающееся с дистальных отделов конечностей. Активные движения пациента способствуют скорейшему восстановлению утраченных двигательных функций.
Частота миоплегических пароксизмов существенно разнится. В тяжелых случаях приступы происходят ежедневно, развивается миоплегический статус либо умеренная мышечная слабость постоянного характера, усиливающаяся по утрам. Это может привести к формированию миопатического синдрома с мышечной гипотрофией, наиболее выраженной в проксимальных отделах. В отдельных случаях пароксизмы носят абортивный характер, когда паралич охватывает лишь ноги, одну конечность, правую или левую половину тела.
Гиперкалиемическая пароксизмальная миоплегия
Встречается намного реже гипокалиемической формы. Характерно аутосомно-доминантное наследование с высокой пенетрантностью. В отдельных семьях пароксизмальная миоплегия прослеживается в 4 поколениях. Лица разных полов заболевают примерно с одинаковой частотой. Типичный возраст манифестации заболевания — первое десятилетие жизни.
Пароксизм миоплегии зачастую провоцируется голодом или возникает в состоянии отдыха после физической нагрузки. Он начинается с парестезий в области лица и конечностей, затем возникает мышечная слабость в дистальных отделах рук и ног, которая быстро распространяется на все мышечные группы, в т. ч. и на лицевую мускулатуру. Процесс сопровождается сухожильной арефлексией и тотальной мышечной гипотонией. Типичны выраженные вегетативные симптомы — сердцебиение, профузный пот, артериальная гипертензия, сильная жажда. Длительность миоплегического пароксизма, как правило, не превышает 1-2 часа.
Нормокалиемическая пароксизмальная миоплегия
Наблюдается крайне редко, в литературе описано лишь 4 семьи с подобной формой миоплегии. Наследуется аутосомно-доминантно, дебют заболевания приходится в основном на первые 10 лет жизни. Нормокалиемическая пароксизмальная миоплегия отличается значительной вариативностью тяжести пароксизмов — от умеренной генерализованной мышечной слабости до полного паралича, включая мышцы лица. У отдельных пациентов отмечается мышечная гипертрофия с формированием атлетического телосложения.
Обычно пароксизм провоцируется переохлаждением или развивается в состоянии покоя после интенсивной физической активности. Характерно медленное нарастание мышечной слабости и еще более замедленное ее уменьшение в конце приступа. Типична большая длительность пароксизмов — от 1-2 дней до нескольких недель.
Диагностика пароксизмальной миоплегии
Типичное течение миоплегических пароксизмов, отсутствие неврологических нарушений в межприступный период, наличие семейного анамнеза — все это позволяет установить диагноз «пароксизмальная миоплегия» без особых затруднений. Сложности возникают при первичном возникновении пароксизма, спорадическом случае заболевания или абортивном характере приступов. Диагноз верифицируется генетиком. Обязательно проводятся консультации невролога и эндокринолога, исключается вторичный характер миоплегии.
Биохимический анализ крови в межприступный период не выявляет каких-либо существенных отклонений. В период пароксизма гиперкалиемической формы миоплегии отмечается понижение уровня калия сыворотки крови до 2 мЭкв/л и ниже (при норме 5,5 мЭкв/л), умеренное снижение содержания фосфора и некоторое повышение сахара крови. При приступе гипокалиемической формы наблюдается значительное увеличение концентрации калия и некоторое понижение сахара крови. Нормокалиемическая пароксизмальная миоплегия отличается отсутствием изменений уровня калия, фосфора и сахара в крови как между приступами, так и в момент пароксизма. Электромиография в момент пароксизма любой из форм миоплегии определяет исчезновение биоэлектрической активности.
С диагностической целью возможно введение пациенту раствора хлорида калия. Развитие миоплегического пароксизма через 20-40 мин свидетельствует о наличии гиперкалиемической формы периодического паралича. В случаях, когда гипокалиемическая пароксизмальная миоплегия протекает с большой частотой приступов и формированием миопатического синдрома, биопсия мышц определяет вакуолярную миопатию. Пароксизмальная миоплегия дифференцируется от миопатии, истерии, восходящего паралича Ландри, нарушений спинального кровообращения, болезни Конна (первичного гиперальдостеронизма).
Лечение пароксизмальной миоплегии
Терапия проводится дифференцированно в зависимости от формы периодического паралича. При гипокалиемической форме для скорейшего окончания пароксизма рекомендован прием 10% р-ра калия хлорида внутрь каждый час, внутривенное введение р-ра калия и магния аспарагината или его прием внутрь. В межприступном периоде для большинства пациентов эффективен спиронолактон. Однако при длительном приеме он способен вызвать нарушения менструального цикла и гирсутизм у женщин, развитие импотенции и гинекомастии у мужчин. В подобных случаях, а также при резистентности к спиронолактону, назначается ацетазоламид. Описана эффективность применения триамтерена (калийсберегающий диуретик). Большое внимание уделяется диете. Рекомендовано снижение количества употребляемой поваренной соли и уменьшение суточного калоража за счет углеводов. Следует ввести в рацион продукты с повышенным содержанием калия — чернослив, курагу, молочные продукты, сыр.
Пароксизмы гиперкалиемической миоплегии хорошо купируются внутривенным введением 40% р-ра глюкозы с 10% р-ром кальция хлорида или инсулином. Некоторые специалисты в области неврологии отмечают положительный эффект ингаляций сальбутамола как для купирования приступов, так и для их предупреждения. В межприступный период назначают гидрохлоротиазид или ацетазоламид. Требуется ограничение в рационе богатых калием продуктов и увеличение потребления поваренной соли и углеводов. Для предупреждения возникновения чувства голода, провоцирующего пароксизм, необходимы более частые приемы пищи.
Нормокалиемическая пароксизмальная миоплегия по причине крайне редкой встречаемости не имеет четко разработанного лечения. Отмечен некоторый эффект ацетазоламида. Рекомендовано увеличение в суточном рационе поваренной соли примерно на 10 г. Симптоматическая пароксизмальная миоплегия требует, в первую очередь, лечения основного заболевания. Параллельно проводится коррекция электролитного баланса.
Прогрессирующий надъядерный паралич ( Прогрессирующая надъядерная офтальмоплегия , Синдром Стила-Ричардсона-Ольшевского )
Прогрессирующий надъядерный паралич — это дегенеративное церебральное заболевание с преимущественным поражением среднего мозга, ядерно-корковых путей, подкорковых образований. Составляющими клинической картины выступают акинетико-ригидная форма паркинсонизма, атаксия, офтальмоплегия, когнитивное снижение, псевдобульбарный синдром. Диагностика осуществляется по клиническим данным, результатам церебральной МРТ и цереброваскулярных исследований. В терапии препаратами выбора являются леводопа, мемантин, антидепрессанты из группы ингибиторов обратного захвата серотонина.
МКБ-10
Прогрессирующий надъядерный паралич (ПНП) — дегенеративное поражение головного мозга неясной этиологии. Наряду с болезнью Альцгеймера, мультисистемной атрофией, кортикобазальной дегенерацией, болезнью Пика, ПНП относится к таупатиям, характеризующимся образованием включений тау-протеина в нейронах и глиальных клетках. Прогрессирующий надъядерный паралич впервые был подробно описан в 1963-64 годах канадскими неврологами Стилом и Ричардсоном в соавторстве с патоморфологом Ольшевским, в честь которых носит название синдром Стила-Ричардсона-Ольшевского. Распространённость заболевания согласно различным информационным источникам варьирует в пределах 1,4-6,4 случая на 100 тыс. населения. Манифестация клинической симптоматики приходится на возрастной период от 55 до 70 лет, с возрастом вероятность развития заболевания увеличивается. Лица мужского пола в большей степени подвержены болезни по сравнению с женщинами.
Причины ПНП
Этиофакторы, запускающие дегенеративные процессы определённой церебральной локализации, остаются неизвестными. Большинство случаев болезни имеют спорадический характер. Отдельные семейные варианты с предположительным аутосомно-доминантным наследованием были выявлены после 1995 года. Молекулярно-генетические исследования показали, что некоторые формы ПНП обусловлены дефектами кодирующего тау-белок гена, локализованного в локусе 17q21.31. Наиболее вероятным представляется мультифакторный механизм возникновения патологии, реализующийся на фоне генетической предрасположенности.
Патогенез
Ведущим патогенетическим механизмом считается дисметаболизм церебральных внутриклеточных белков, сопровождающийся избирательной агрегацией отдельных белков (тау-протеина, убиквитина) в определённых группах мозговых клеток. Патологические включения нарушают жизнедеятельность нейронов, запускают процесс деградации и запрограммированной гибели (апоптоза). Дегенеративные изменения носят селективный характер, распространяются преимущественно на средний мозг, зубчатые мозжечковые ядра и подкорковые структуры: черную субстанцию, бледный шар, таламус, ретикулярную формацию, субталамическое ядро. В меньшей степени поражается кора префронтальных и височных зон.
Патоморфологическая картина ПНП представлена наличием нейрофибриллярных клубочков, глиальных включений, нитевидных белковых образований в нейронах указанных церебральных структур. Макроскопически определяется атрофия среднего мозга с существенным уменьшением его сагиттального размера. Поражение среднего мозга обуславливает надъядерный паралич глазодвигательной мускулатуры, дегенерация кортико-бульбарных трактов — псевдобульбарные проявления. Нейрохимические исследования выявляют пониженную концентрацию дофамина в стриатуме, лежащую в основе паркинсонического симптомокомплекса.
Симптомы ПНП
Прогрессирующий надъядерный паралич характеризуется неспецифичным клиническим дебютом. Симптоматика этого периода представлена непривычной утомляемостью, сниженной работоспособностью, цефалгиями, головокружением, пониженным настроением, сужением круга интересов, нарушениями сна, включающими бессонницу ночью и гиперсомнию днём. В последующем присоединяются симптомы акинетико-ригидного паркинсонизма. Постуральный тремор у большинства пациентов отсутствует. Мышечная ригидность выражена преимущественно в аксиальной мускулатуре — мышцах, идущих вдоль шейного отдела позвоночника, соединяющих его с черепом. Больные жалуются на скованность в шее, спине. Повышение тонуса в задних мышцах шеи приводит к типичному «горделивому» положению головы пациента. Характерна паркинсоническая атаксия, обусловленная расстройством координации положения туловища и нижних конечностей относительно центра тяжести. Затруднения в поддержании равновесия в процессе ходьбы приводят к частым падениям назад.
Отличительной особенностью ПНП выступает офтальмоплегия, возникающая в среднем спустя 2-3 года от дебюта заболевания. На фоне замедленного движения глазных яблок происходит паралич взора в вертикальной плоскости, пациент не может опустить глаза вниз. Из-за редкого моргания больной ощущает дискомфорт, жжение в глазах. Возможны расплывчатость зрения, расстройство конвергенции, блефароспазм. Прогрессирующий надъядерный офтальмопарез сопровождается ограничением взора вниз и вверх, со временем может приводить к глазодвигательным нарушениям в горизонтальной плоскости. При развитии полной офтальмоплегии формируется ретракция верхних век, что придаёт лицу удивлённое выражение.
В клинической картине ПНП относительно рано возникают псевдобульбарные проявления: дизартрия, дисфагия, насильственный плач или смех. Происходят изменения личностно-эмоциональной сферы, больные становятся замкнутыми, апатичными, демотивированными, безразличными. Когнитивные нарушения в большинстве случаев присоединяются в разгаре болезни, в 10-30% случаев — на стадии дебюта. Характерно интеллектуальное снижение, расстройства абстрактного мышления и памяти, зрительно-пространственная апраксия, элементы агнозии. Деменция наблюдается у 60% пациентов с 3-летним стажем заболевания.
Осложнения
В начальном периоде падения больного без возможности скоординировать свои движения приводят к ушибам и переломам. Спустя несколько лет прогрессирующий олигобрадикинетический синдром приковывает пациентов к постели. При отсутствии должного ухода обездвиженность опасна развитием контрактур суставов, пролежней, застойной пневмонии. Прогрессирующий псевдобульбарный паралич обуславливает попёрхивание пищей с риском асфиксии, аспирационной пневмонии. Ночные апноэ могут стать причиной внезапной смерти во сне. Серьёзным осложнением является присоединение интеркуррентных инфекций (пневмонии, цистита, пиелонефрита), поскольку на фоне сниженного иммунитета существует высокий риск развития сепсиса.
Диагностика
Вероятными ранними критериями ПНП являются начало после 40-летнего возраста, прогрессирующий характер, парез горизонтального взора, выраженная постуральная неустойчивость с эпизодами падений. Постановка достоверного диагноза возможна при наличии гистологически подтверждённых патогномоничных для ПНП изменений в тканях мозга. Перечень необходимых диагностических исследований включает:
- Осмотр невролога. В неврологическом статусе ведущим синдромом является симметричная олигобрадикинезия. Наблюдается гипомимия, ретроколлис (патологическая установка шеи), парез вертикального взора, симптомы орального автоматизма, повышение сухожильных рефлексов. Выражена постуральная неустойчивость.
- Нейропсихологическое тестирование. Проводится психиатром, нейропсихологом с использованием специальных тестов, заданий (шкалы MMSE, MоCА, теста рисования часов). Требуется для оценки наличия и степени выраженности когнитивного снижения. Надъядерный паралич проявляется замедленным мышлением, быстрой истощаемостью, умеренной выраженностью интеллектуальных нарушений.
- МРТ головного мозга. Выявляет расширение III желудочка, атрофические изменения среднего мозга, базальных ганглиев, премоторных зон лобной коры и височных областей. Позволяет исключить внутримозговую опухоль, энцефалит, рассеянный склероз, инсульт.
- Оценку церебральной гемодинамики. Данные о кровоснабжении мозга могут быть получены путём дуплексного сканирования, УЗДГ, МРТ сосудов. Необходимы для исключения дисциркуляторной энцефалопатии, сосудистого паркинсонизма, сосудистой деменции.
Дифференциальная диагностика осуществляется с болезнью Паркинсона, вторичным паркинсонизмом травматической, инфекционной, токсической, сосудистой этиологии, деменциями альцгеймеровского типа, поздней формой нейроакантоцитоза. От классической болезни Паркинсона надъядерный паралич отличается симметричностью паркинсонизма с момента его появления, быстрым развитием когнитивных расстройств, офтальмоплегией, ретроколлисом, выраженной атаксией, малым эффектом дофаминергической терапии. Достоверно дифференцировать прогрессирующий надъядерный паралич от прочих таупатий можно по особенностям патоморфологических изменений.
Лечение ПНП
Эффективная терапия, способная остановить прогрессирующий дегенеративный процесс, пока не найдена. Осуществляется симптоматическое лечение, направленное на облегчение состояния пациента. Проведенные фармакотерапевтические исследования не сопровождались плацебо-контролем, слабо доказывают эффективность медикаментозной терапии. В лечении когнитивных нарушений возможно применение мемантина, ингибиторов ацетилхолинэстеразы, для коррекции психоэмоциональной сферы — антидепрессантов с психоактивирующим действием (флуоксетина, пароксетина).
Большинство неврологов считают необходимым назначение стартовой дофаминергической терапии. У половины больных наблюдается определённое облегчение состояния на фоне приёма препаратов леводопы, однако данный эффект длится не более двух лет. Противопаркинсонические фармпрепараты прочих групп (ингибиторов МАО, агонистов дофаминовых рецепторов, ингибиторов КОМТ) не показали своей эффективности.
Прогноз и профилактика
При надъядерном параличе наблюдается безостановочное прогрессирование симптоматики. Проводимая терапия не оказывает существенного эффекта на течение болезни. Продолжительность жизни пациентов колеблется в пределах 5-15 лет. Летальный исход обусловлен интеркуррентными инфекциями, затяжным апноэ сна, аспирационной пневмонией. В связи с отсутствием ясного понимания этиологии и патогенеза нозологии разработка профилактических мероприятий не представляется возможной, исследования заболевания и методов его лечения продолжаются.
1. Клинические особенности надъядерного паралича/ Валикова Т.А., Алифирова В.М., Пугаченко Н.В., Цыренжапова Р.Б., Бичик А.Б.// Бюллетень сибирской медицины - 2009. - №3 (2).
2. Cлучай прогрессирующего надъядерного паралича с кортикобазальным синдромом/ Федотова Е.Ю., Чечеткин А.О., Иванова-Смоленская И.А., Иллариошкин С.Н.// Атмосфера. Нервные болезни. - 2009 - №2.
3. Трудности диагностики прогрессирующего надъядерного паралича/ Ситкали И.В., Раздорская В.В.// Бюллетень медицинских интернет-конференций. - 2015 - Т.5, №4.
4. Трудности дифференциальной диагностики прогрессирующего надъядерного паралича и болезни Паркинсона/ Магжанов Р.В., Давлетова А.И., Ибатуллин Р.А., Туник В.Ф., Идрисова Р.Ф., Бахтиярова К.З.// Анналы клинической и экспериментальной неврологии. - 2016.
Гипокалиемический периодический паралич
Гипокалиемический периодический паралич - группа генетически обусловленных миоплегий, характеризующихся пароксизмальными нарушениями работы мышц, сопровождающихся снижением уровня калия. Их симптомами являются внезапные приступы мышечной слабости вплоть до почти полного паралича, в основном поражается мускулатура конечностей, иногда с вовлечением дыхательных мышц, что способно создавать угрозу жизни. Диагностика производится на основании клинической картины, измерения уровня калия в период приступов, изучения наследственного анамнеза пациента и генетических исследований. Специфического лечения не существует, возможно купирование приступов препаратами калия и профилактика их развития калийсберегающими средствами.
Гипокалиемический периодический паралич (в некоторых источниках - болезнь Вестфаля) - несколько сходных по своим клиническим проявлениям наследственных заболеваний, обусловленных дефектами ионных каналов мышечных клеток. Впервые подобное состояние было описано в 1884 году русским врачом И. В. Шахновичем, который обнаружил классический вариант заболевания. В дальнейшем были обнаружены и его иные формы, которые, помимо генетически-молекулярных параметров и клинических проявлений, различаются между собой по своему географическому распространению. В целом, гипокалиемический периодический паралич примерно в 3 раза чаще встречается у мужчин, чем у женщин, общая встречаемость составляет примерно 1:100000. Считается, что механизм наследования всех форм этого заболевания является аутосомно-доминантным, однако имеются указания и на наличие рецессивных семейных форм. Обычно при своевременно поставленном диагнозе приступы удается легко купировать, но иногда они могут угрожать жизни больного по причине вовлечения дыхательной мускулатуры.
Причины гипокалиемического периодического паралича
Общим для всех типов гипокалиемического периодического паралича является нарушение функционирования ионных каналов миоцитов скелетных мышц, обусловленное различными мутациями. При помощи методов современной генетики удалось идентифицировать три гена, дефекты которых приводят к такому состоянию. За классический вариант заболевания ответственны мутации гена CACNL1A3, расположенного на 1-й хромосоме. Он кодирует особый белок мембран миоцитов - альфа-субъединицу дегидроптерин-чувствительного кальциевого канала, из-за чего функционирование этого ионного пути нарушается. Патогенез нарушений при этой форме гипокалиемического периодического паралича изучен недостаточно, предполагается, что изменение проницаемости клеточных мембран для ионов кальция вносит дисбаланс во всю систему ионных взаимодействий миоцитов. В результате время деполяризации мышечных клеток существенно удлиняется, что может приводить к приступам слабости.
Второй генетически-молекулярный тип заболевания обусловлен миссенс-мутациями гена SCN4A, расположенного на 17-й хромосоме. Также, как и CACNL1A3, этот ген кодирует один из компонентов ионного канала миоцитов, только другого типа - альфа-субъединицу натриевого канала. Данная разновидность гипокалиемического периодического паралича сопровождается не только приступами слабости и снижением уровня калия в крови, но и в ряде случаев низкой концентрацией инсулина и аномальной реакцией миоцитов на стимуляцию этим гормоном. Возможно, это связано с тем, что эффекты инсулина во многом обусловлены именно различными типами транспорта ионов натрия через мембрану клеток, и нарушение этого процесса приводит к различным сбоям. Некоторыми исследователями описаны случаи аутосомно-рецессивного наследования мутаций гена SCN4A.
Третий клинический тип гипокалиемического периодического паралича вызывается мутациями гена KCNE3, локализованного на 11-й хромосоме. Продуктом его экспрессии является протеин калиевых каналов миоцитов скелетных мышц, поэтому при дефекте его структуры напрямую возникают нарушения в транспорте ионов калия. Интересно, что аналогичные мутации в некоторых семьях сопровождались гиперкалиемическим периодическим параличом, что может свидетельствовать об аллельности этих состояний. В отличие от других типов заболевания, при мутациях гена KCNE3 нередко затрагивается и миокард, что выражается в нарушении сердечного ритма на высоте приступа мышечной слабости и паралича.
Механизмом развития гипокалиемии при всех трех формах заболевания является изменение проницаемости мембран миоцитов, в результате чего калий легче, чем у здоровых людей, начинает проникать в клетки, исчезая при этом из межклеточного пространства и плазмы крови. В результате этого изменяется трансмембранный потенциал (обеспечиваемый разной концентрацией ионов натрия и калия снаружи и внутри клетки), и процесс деполяризации мембраны сильно осложняется и удлиняется. Так как именно деполяризация играет центральную роль в развитии потенциала действия и мышечного сокращения, нарушения этого процесса клинически выражаются в развитии приступа гипокалиемического периодического паралича.
Классификация гипокалиемического периодического паралича
На сегодняшний день известно три формы гипокалиемического периодического паралича, которые отличаются между собой этиологией, клинической картиной, географическим распространением. Встречаемость каждого типа в отдельности не определена, поэтому используют усредненное значение.
- Классический тип гипокалиемического периодического паралича - обусловлен мутацией гена CACNL1A3 и вызванного этим дефектом одного из кальциевых каналов. Распространен повсеместно, четкой привязки к конкретному региону планеты у этой формы не наблюдается.
- Дистальный почечный тубулярный ацидоз - форма гипокалиемического периодического паралича, которая вызвана мутацией гена SCN4A, наиболее распространена в странах Юго-Восточной Азии (Тайланд, Индонезия). Характерной особенностью этого типа, помимо поражения мышц, являются выраженные метаболические расстройства, патологии костной системы (остеомаляции) и почек.
- Периодический тиреотоксический паралич - обусловлен мутацией гена KCNE3, особенно распространен в Азии и Северной Америке. На высоте приступа могут возникать нарушения сердечного ритма, однако летальные случаи встречаются достаточно редко. Иногда этот тип заболевания ошибочно считают вторичной патологией, обусловленной не генетическими нарушениями, а поражением щитовидной железы.
По мнению некоторых врачей-генетиков, вышеуказанная классификация с привязкой конкретной формы гипокалиемического периодического паралича к определенным генам не совсем корректна. Они указывают, что имеются случаи, когда, например, мутации гена KCNE3 проявляли себя как первый клинический тип заболевания. Данная теория требует всестороннего изучения и подтверждения, на сегодняшний день нет четких доказательств ее правильности.
Симптомы гипокалиемического периодического паралича
Ведущим симптомом всех форм гипокалиемического периодического паралича являются пароксизмальные приступы мышечной слабости разной продолжительности, способные поражать различные группы мышц. Классический тип заболевания характеризуется началом в возрасте от 3-х до 22-х лет и вовлечением сразу большого объема мускулатуры - нижних и верхних конечностей, туловища, шеи. Мимические и жевательные мышцы, как правило, не поражаются, в тяжелых случаях может наблюдаться вовлечение дыхательной мускулатуры - без своевременной медицинской помощи это способно привести к внезапному летальному исходу. Чаще всего приступы классического гипокалиемического периодического паралича возникают ночью или утром, сопровождаются адинамией (из-за большого объема вовлеченных мышц), провоцирующим фактором может выступить переохлаждение, значительные физические нагрузки накануне, инфекционные заболевания, болезненные менструации. Также имеются указания, что прием чрезмерных количеств поваренной соли (например, соленых продуктов) также способен вызвать приступ мышечной слабости.
Длительность и частота приступов может быть различна, даже у одного больного наблюдается сильная вариация в этом отношении - подвижность мышц может в одном случае вернуться через час, а в другом - через сутки. Помимо этого во время приступа могут наблюдаться и вегетативные нарушения - гипергидроз (ладонный, стопный), тошнота, рвота. Характерны колебания артериального давления, что может усугубить состояние больного. С возрастом эпизоды классического гипокалиемического периодического паралича становятся все реже, описано множество клинических случаев, когда после 50-55 лет они пропадали у больного вовсе.
Второй тип гипокалиемического периодического паралича - дистальный почечный тубулярный ацидоз - является аллельной разновидностью гемолитической анемии и сфероцитоза. Приступы мышечной слабости в основном поражают конечности (преимущественно проксимальные отделы), иногда может наблюдаться вовлечение дыхательной мускулатуры. Длительность эпизодов невелика - около часа, но их развитие абсолютно спонтанно, так как выявить достоверные провоцирующие факторы до сих пор не удалось. Помимо мышечных расстройств, при этом типе гипокалиемического периодического паралича нередко развивается ацидоз, симптомами которого могут выступать головная боль, тошнота. Также характерны остеомаляции, которые приводят к частым переломам костей - это служит дополнительным симптомом именно этой формы заболевания.
Третья форма гипокалиемического периодического паралича, или периодический тиреотоксический паралич, обнаруживает себя в возрасте от 20 до 40 лет, длительность приступов мышечной слабости составляет от 1-го до 20-ти часов. Преимущественно поражается мускулатура ног, в меньшей степени рук; замечено, что чем интенсивней работает та или иная группа мышц, тем больший риск ее вовлечения в эту форму заболевания. По этой причине развитию эпизода паралича предшествует физическая нагрузка, после которой и начинают проявляться симптомы заболевания. Кроме того, в отношении этой разновидности гипокалиемического периодического паралича выявлена четкая сезонность - большинство приступов развивается в период с мая по октябрь. Достоверных объяснений этому нет, предполагается, что развитие мышечной слабости облегчается на фоне потери калия с потом в летние месяцы. Также на высоте эпизода может возникать нарушение сердечного ритма, дыхательная мускулатура практически никогда не вовлекается в патологический процесс. Также наблюдаются нарушения со стороны щитовидной железы с развитием симптомов тиреотоксикоза - тремора, похудения, экзофтальма.
Помимо вышеуказанных провоцирующих факторов для каждой формы, привести к развитию приступа могут иные обстоятельства - особенно те, которые способствуют снижению уровня калия в плазме крови. К ним относят некоторые эндокринные заболевания, прием богатой углеводами пищи, инъекции инсулина. Среди других лекарственных средств, способных спровоцировать приступ гипокалиемического периодического паралича, можно выделить адренокортикотропный гормон (АКТГ), минералокортикоиды, калийнесберегающие диуретики. Поэтому лицам, страдающим от этого заболевания, следует с осторожностью назначать указанные препараты и при их использовании постоянно контролировать концентрацию ионов калия в плазме крови.
Диагностика гипокалиемического периодического паралича
Выявление гипокалиемического периодического паралича производится при помощи осмотра пациента в период приступов, электромиографических исследований, биохимического анализа крови, генетических методик и в некоторых случаях - биопсии мышц. Результаты всех этих исследований могут несколько отличаться в зависимости от клинической формы заболевания, что позволяет идентифицировать их даже без использования молекулярно-генетической диагностики. Общими для всех форм патологии проявлениями служат мышечная слабость, резкое снижение или полное отсутствие сухожильных рефлексов на высоте приступа, отсутствие реакции мышц на любые раздражители, в том числе и электрический ток. В период между эпизодами мышечной слабости диагностировать гипокалиемический периодический паралич нередко можно только генетическими исследованиями, так как никаких других признаков заболевания не будет выявляться. Даже гистологическое изучение биоптата мышечной ткани в ряде случаев не обнаруживает никаких патологических изменений.
При приступе мышечной слабости биохимический анализ крови выявляет выраженную гипокалиемию, при второй форме заболевания она также может сопровождаться снижением уровня инсулина, ацидозом и гиперхлоремией. Также наблюдается незначительное повышение уровня креатинфосфокиназы, однако это является непостоянным признаком, нередко проявляющимся лишь на самом пике приступа гипокалиемического периодического паралича. Анализ мочи, выполненный в период эпизода мышечной слабости, часто показывает значительное снижение рН. На электрокардиограмме могут выявляться нарушения сердечного ритма (экстрасистолы, аритмии), а также другие признаки гипокалиемии - депрессия сегмента ST и сглаживание зубца Т. При осмотре пациента может быть обнаружено затрудненное дыхание - причиной этого может стать либо вовлечение в патологический процесс дыхательной мускулатуры, либо гипервентиляция при ацидозе (при втором типе гипокалиемического периодического паралича).
Биопсия мышц, выполненная в период между приступами заболевания, может не выявить никаких изменений - лишь при частых и тяжелых эпизодах возможно развитие признаков атрофии и дегенерации мышечных волокон. При гистологическом изучении тканей, полученных во время приступа, можно выявить развитие вакуолей в миоцитах, наличие расширенных участков эндоплазматической сети, неодинаковую толщину мышечных волокон. Генетическая диагностика сводится к секвенированию последовательностей ассоциированных с гипокалиемическим периодическим параличом генов для выявления дефектов и мутаций. Возможна пренатальная диагностика посредством амниоцентеза или биопсии ворсин хориона. Дифференцировать это заболевание следует с приобретенными формами гипокалиемии, некоторыми миопатиями.
Лечение гипокалиемического периодического паралича
Специфической или этиотропной терапии гипокалиемического периодического паралича не существует, все лечебные мероприятия сводятся к купированию приступов мышечной слабости путем восстановления ионного баланса организма и профилактике дальнейших эпизодов. При развитии паралича назначают хлорид калия или другую соль этого элемента, в зависимости от выраженности симптомов его могут назначать как внутрь, так и парентерально (струйно или капельно). Желательно избегать введения солей калия вместе с глюкозой, особенно при низкой концентрации раствора - это может еще больше понизить уровень данного иона в плазме и усугубить состояние больного. При введении лекарственных средств необходимо постоянно контролировать сердце (ЭКГ) и концентрацию калия в плазме во избежание гиперкалиемии.
Профилактическое лечение включает в себя назначение препаратов калия с ацетазоламидом - в ряде случаев это значительно уменьшает частоту и выраженность приступов гипокалиемического периодического паралича. В этом случае необходим контроль концентрации ионов калия в плазме крови, а также работы почек, так как ацетазоламид способен вызывать образование почечных камней. У некоторых больных отсутствует реакция на применение этого препарата, в такой ситуации его можно заменить спиронолактоном или триамтереном. Немаловажную роль в терапии гипокалиемического периодического паралича играет соблюдение низкоуглеводной диеты, употребление богатых калием продуктов, выполнение специальных физических упражнений для предотвращения хронической мышечной слабости.
Прогноз и профилактика гипокалиемического периодического паралича
Прогноз гипокалиемического периодического паралича во многом зависит от выраженности симптомов. При вовлечении дыхательной мускулатуры и нарушениях сердечного ритма возможна внезапная смерть на высоте приступа. Но в большинстве случаев прогноз относительно жизни благоприятный, с возрастом частота и сила эпизодов снижается. Возможно развитие хронической мышечной слабости по причине частых приступов, поэтому необходимо их своевременно купировать, а также выполнять все требования по профилактике их развития. Помимо лекарственных мер, профилактика осуществляется правильно сбалансированным рационом с низким содержанием углеводов и включением в него продуктов, богатых калием. Желательно избегать значительных физических нагрузок, а также применения гормональных или мочегонных средств без назначения и контроля врача.
Периодический паралич
Периодический паралич (ПП) является редким генетическим заболеванием. Он проявляется внезапными приступами кратковременной мышечной слабости, скованности или паралича. Подобные атаки могут затронуть все тело или только одну или две конечности.
Существует несколько форм ПП. Все они связаны с дефектами ионных каналов. Ионные каналы - это шлюзы, которые позволяют заряженным частицам минералов (ионам), таких как натрий и калий, проникать в клетки и выходить из них. Потоки ионов являются центральной частью работы мышц. При ПП ионные каналы выходят из строя, и мышечные клетки перестают работать правильно.
Приступы ПП могут начаться в детстве или в зрелом возрасте. Они могут возникнуть после тяжелых физических упражнений или действия других триггеров. В зависимости от имеющейся формы ПП симптомы могут быть как слабыми, так и тяжелыми, могут длиться как минуты, так и дни. Иногда болезнь прогрессирует и со временем вызывает постоянное повреждение мышц.
Основные формы ПП:
- Гипокалиемический ПП. Приступы являются результатом низкого уровня калия в крови. Триггеры могут включать физические упражнения, употребление продуктов с большим количеством сахара и крахмала (углеводов), солодки, стресс, низкие температуры и прием некоторых лекарств. Это самая распространенная форма ПП.
- Гиперкалиемический ПП. Приступы являются результатом высокого уровня калия в крови. Триггеры могут включать физические упражнения, голодание, стресс, простуду и прием некоторых лекарств.
- Тиреотоксический ПП. Приступы являются результатом высокого уровня гормонов щитовидной железы. Обычно при этом присутствуют проблемы со щитовидной железой. Триггеры могут включать физические упражнения, прием пищи с большим количеством углеводов и стресс. Эта форма встречается в основном у мужчин, особенно азиатского происхождения.
- Синдром Андерсена-Тавила. Приступы обусловлены колебанием уровня калия в крови. Триггеры могут включать физические упражнения, стресс и прием определенных лекарств. Многие люди с этой формой ПП имеют определенный набор черт лица. К ним относятся широкий лоб, широко расставленные глаза, низко посаженные уши и маленький подбородок.
Большинство форм ПП поражают скелетные мышцы, т. е. те, которые управляют движениями. Синдром Андерсена-Тавила может поражать не только скелетные мышцы, но и мышцы сердца, делая эту форму ПП более опасной, чем большинство других форм.
Что вызывает периодический паралич?
Все формы ПП являются результатом генетических патологий. Эти патологии приводят к дефектам ионных каналов. Ионные каналы управляют тем, как заряженные минералы (ионы), такие как калий, натрий и кальций, попадают в мышечные клетки и покидают их.
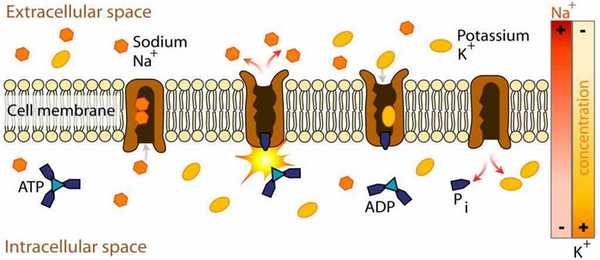
Каждая мышечная клетка имеет покрытие или мембрану, которая защищает внутреннюю часть клетки. Когда нерв требует от мышечной клетки сокращаться, он посылает химический сигнал, который открывает ворота, чтобы позволить ионам натрия проникнуть в клетку. Поток заряженных ионов изменяет электрический заряд внутри клетки, что вызывает волну тока через мышечное волокно. Ионы кальция выходят из резервуаров в клетках и заставляют мышечные волокна сокращаться, затем калиевые каналы открываются, отправляя ионы калия из клетки. Это вызывает сокращение мышцы.
В ПП ионные каналы имеют дефекты, которые могут нарушить процесс. В этом случае мышечные клетки не могут сжиматься или расслабляться в ответ на нервные сигналы.
Есть десятки различных дефектов, которые могут вызвать ПП. Они затрагивают натриевые, кальциевые или калиевые ионные каналы. Есть также много нераспознанных дефектов, приводящих к ПП. В большинстве случаев для того, чтобы получить ПП, достаточно, чтобы только один родитель имел это заболевание.
Каковы симптомы периодического паралича?
Симптомы в некоторой степени зависят от формы ПП. Впервые пациент может заметить симптомы в детстве или в зрелом возрасте. Приступы могут случаться часто или быть редкими. При некоторых формах ПП количество атак уменьшается с возрастом. Как правило, у пациентов с ПП имеются следующие симптомы:
- приступы мышечной слабости длительностью от нескольких минут до нескольких дней;
- мышечные боли после физической нагрузки;
- мышечные спазмы;
- ощущение покалывания в мышцах;
- постоянная слабость, которая может сохраняться и вне приступов.
Различные формы ПП могут вызывать следующие проявления:
- Гипокалиемический ПП часто начинается в позднем детском или подростковом возрасте. Средний возраст от 5 до 35 лет. Приступы слабости скелетных мышц могут длиться от пары часов до суток. Обычно они происходят ночью или утром. Во время самых тяжелых эпизодов человек вообще не в состоянии двигаться. Приблизительно после 50 лет заболевание приводит к постоянной слабости, которая постепенно усиливается, особенно в бедрах.
- Гиперкалиемический ПП часто начинается в возрасте 10 лет. Приступы слабости скелетных мышц длятся в среднем от 30 минут до 4 часов. Приступы, как правило, частые, но менее серьезные, чем при других формах ПП. Когда пациент становится старше, атаки случаются реже, однако может развиться постоянное повреждение мышц, которое будет постепенно прогрессировать.
- Тиреотоксический ПП начинается в возрасте от 20 до 40 лет. Приступы случаются от нескольких раз в год до нескольких раз в неделю, их продолжительность может измеряться часами или днями. Этой форме ПП также могут сопутствовать симптомы, связанные с щитовидной железой, такие как беспокойство, потливость, потеря веса и аномальное ощущение сердцебиения. У некоторых людей с тиреотоксическим ПП симптомы заболевания щитовидной железы отсутствуют, особенно на ранних стадиях.
- Синдром Андерсена-Тавила обычно начинается в возрасте до 18 лет. Приступы длятся от 1 до 36 часов. Эта форма может вызвать сердечную аритмию, потому что она наряду со скелетными мышцами затрагивает сердечную мышцу.
Симптомы отличаются в зависимости от формы ПП и конкретного генного нарушения, которое его вызывает.
Как диагностируется периодический паралич?
Вначале врач изучит историю болезни. Он (она) спросит о недавних симптомах, прошлых проблемах со здоровьем и семейной истории болезни. Затем врач проведет медицинский осмотр и оценит состояние мышц. Могут понадобиться:
- анализ крови на уровень калия во время приступа;
- анализы крови для измерения уровня других минералов и газов крови;
- анализы крови на известные генетические дефекты, вызывающие ПП;
- исследования нервной проводимости и электромиография для измерения электрической активности мышц;
- электрокардиограмма (ЭКГ) для проверки электрической активности сердца;
- анализы крови на содержание гормонов щитовидной железы.
Вначале можно обратиться к своему основному врачу, но затем следует обратиться к неврологу, который специализируется на заболеваниях, подобных ПП.
Как лечится периодический паралич?
Лечение направлено на уменьшение количества и тяжести приступов. Методы управления ПП могут включать:
- Тщательный контроль потребления калия с помощью диеты и добавок.
- Внутривенный прием калия, если тяжелые симптомы вызваны гипокалиемическим ПП.
- Контроль углеводов в рационе.
- Контроль функции щитовидной железы, что очень важно при тиреотоксическом ПП.
- Получение помощи от диетолога. Этот специалист поможет составить диету, которая уменьшит атаки, изменяя количество углеводов или минералов.
- Прием лекарственных средств, таких как ацетазоламид. Следует соблюдать осторожность, так как в некоторых случаях этот препарат может усилить ПП.
- Прием других лекарств, таких как ингибиторы карбоангидразы или калийсберегающие диуретики.
- Отказ от определенных видов анестезии из-за возможных осложнений.
- Прием антиаритмических лекарственных средств или имплантация кардиостимулятора при синдроме Андерсена-Тавила.
- Ограничение влияния триггеров и поддержание физической активности на умеренном уровне.
- Прием дихлорфенамида. Этот лекарственный препарат может уменьшить количество приступов при гипокалиемическом ПП.
Может потребоваться частый мониторинг уровня калия в крови.
Комбинация контролируемой диеты, лекарств и изменений в образе жизни обычно помогает управлять ПП.
Ключевые моменты
ПП - это редкое заболевание, которое вызывает внезапные приступы временной мышечной слабости, скованности или паралича.
ПП - это генетическое заболевание, полученное от родителей. Наследование является доминантным, что означает, что у вас есть шанс получить его, если хотя бы один из родителей имеет генетический дефект, определяющий возникновение ПП.
Эпизоды ПП могут начаться в детстве или в зрелом возрасте.
Приступы могут быть легкими или серьезными, они могут длиться минуты или дни.
В зависимости от формы ПП приступы могут возникать из-за низкого или высокого уровня калия в крови, физических нагрузок, стресса, простуды, богатой углеводами пищи, голодания, действия некоторых лекарств или высокого уровня гормонов щитовидной железы.
Синдром Андерсена-Тавила может затрагивать сердечную мышцу, вызывая нерегулярное сердцебиение (аритмию).
С возрастом повреждение мышц может приобрести постоянный характер.
Для лечения ПП можно использовать комбинацию диеты, лекарственной терапии и изменения образа жизни.
Читайте также:
- Аутологичная трансплантация гемопоэтических стволовых клеток (ауто-ТГСК) при остром миелобластном лейкозе (ОМЛ)
- Диагностика клебсиел. Микробиологическая диагностика клебсиелл. Выявление клебсиел. Лечение клебсиеллезов. Профилактика клебсиеллезов.
- Прогноз холестеатомы. Лечение хронического эпитимпанита
- Врожденный анкилоблефарон: причины, диагностика, лечение
- Протез и железы твердого неба. Влияние протеза на железы протезного ложа