Врожденные миопатии у ребенка
Добавил пользователь Владимир З. Обновлено: 21.01.2026
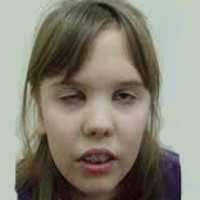
Миастения является аутоиммунным заболеванием, которое проявляется необычной для ребенка утомляемостью и преходящей слабостью мышц. Данной болезни чаще подвержены девочки, средний возраст больных составляет 7 лет. Однако клинически подтверждено, что недуг может поражать и подростков (до 15% всех случаев), и взрослых.
Каковы причины миастении?
Приобретенная форма часто развивается на фоне стресса, нарушений функции иммунной системы, вирусов. Из всего числа больных данной формой на детский возраст приходится 1-3%.
Течение и особенности заболевания
Если у матери имеется миастения, то существует 10-20% шанс рождения ребенка с этим недугом. Однако миастенический синдром в этом случае имеет преходящую форму (длится от нескольких дней до 1,5 месяцев).
У новорожденных болезнь проявляется следующим образом:
- слабый крик;
- гипотония мышц;
- трудности при сосании и дыхании.
Также могут развиваться:
- опущение (птоз) одного из органов;
- нарушение движения глаз;
- анемия;
- угнетение глубоких рефлексов;
- затруднение глотания.
Врожденная миастения у детей раннего возраста может провоцировать следующие типы расстройств:
- общая мышечная слабость с нарушением дыхания и функции сердца, а также без данных проявления;
- локальная мышечная слабость (лицо и глотка без нарушения дыхания и с ним; глаза);
- скелетно-мышечная слабость с нарушением дыхания и без.
Миастения классифицируется по нескольким признакам:
- возраст больного (неонатальная, юношеская, болезнь взрослых);
- наличие антител (серопозитивная, серонегативная);
- клиническая картина (преходящая с полным регрессом, непрогрессирующая в течение нескольких лет, прогрессирующая, злокачественная со стремительным нарастанием нарушений);
- степень нарушений (общая, локальная);
- тяжесть протекания (легкая, средняя, тяжелая);
- полнота восстановления после введения антител (полная, неполная, низкая);
- наличие ухудшения важных для жизни функций организма.
Симптомы у детей
Для малышей характерны следующие проявления заболевания:
- птоз органа;
- недостаточный объем движения глаз;
- снижение жевательной функции;
- двоение в глазах;
- нарушение мимики;
- трудности при глотании;
- необычная утомляемость верхних конечностей, шеи, мышц таза;
- неподвижный взгляд, опущенные веки, необычное выражение лица;
- речевые расстройства;
- гнусавость. Источник:
М.Л. Чухловина, Н.П. Шабалов, Н.В. Цинзерлинг
Особенности патогенеза, клиники и диагностики миастении в детском возрасте
// Педиатрия, 2006, №3, с.90-94
В основном болезнь начинается с одного симптома, но к нему присоединяются другие довольно стремительно. Первые несколько месяцев процесс распространяется на весь организм. Также встречаются локальные формы, которые у детей протекают тяжелее, чем у взрослых.
Для общей формы характерно вовлечение мышц туловища, шеи, конечностей, быстрая утомляемость и слабость, усиливающиеся после нагрузки.
Локальные формы подразделяются по пораженным отделам:
- глазное расстройство влечет нарушение движений глаз, косоглазие, одно- или двухсторонний птоз, двоение в глазах. Данные симптомы усиливаются к вечеру, могут снижаться после отдыха;
- скелетно-мышечное и глоточно-лицевое расстройство выражается в нарушение функции сердца и дыхания;
- отдельно глоточно-лицевая форма вызывает нарушение мимики, слабость языка, мускул гортани, что выражается в речевых и глотательных нарушениях.
Методы диагностики
- ЭНМГ.
- Серологическое исследование для определения наличия антител.
- Оценка внешней клинической картины.
- Клинические пробы.
- Дифференциальная диагностика с целью исключения болезней, которые имеют среди проявления миастенический синдром. Источник:
А.И. Смолин
Современные аспекты клиники и диагностики и миастении
// Сибирский медицинский журнал, 2013, №3, с.12-15
Лечение миастении у детей
Для лечения детей применяются консервативные и радикальные методы. К консервативным относится применение ряда препаратов. Они подбираются индивидуально на основе клинической картины и тяжести заболевания.
В ходе же хирургического вмешательства удаляется вилочковая железа. Именно она вызывает миастению в 70-80% случаев. Такое лечение показано при средней или тяжелой форме заболевания.
Для родителей крайне важно как можно скорее обратиться к специалисту - неврологу - для своевременной диагностики и назначения терапии. Это связано с тем, что при малой продолжительности миастении прогноз гораздо лучше, чем в том случае, когда болезнь длится более пяти лет.
В медицинском центре «СМ-Клиника» Вас готовы принять одни из лучших детских врачей Санкт-Петербурга. Запишитесь на прием как можно скорее, и Ваш ребенок будет здоров и счастлив.
Maria F. Finnis and Sandeep Jayawant. Juvenile Myasthenia Gravis: A Paediatric Perspective // Autoimmune Dis. 2011; 2011: 404101.
Пицуха Светлана Анатольевна
Clinic
Врач высшей квалификационной категории
Информация в статье предоставлена в справочных целях и не заменяет консультации квалифицированного специалиста. Не занимайтесь самолечением! При первых признаках заболевания необходимо обратиться к врачу.
Немалиновая миопатия
Немалиновая миопатия - группа генетически разнородных наследственных миопатий, общим патогистологическим проявлением которых является образование в мышечной ткани нитевидных структур, что нашло свое отражение в названии патологии (от греческого Nema - нить). Симптомы различных форм состояния могут значительно различаться по своей выраженности - возможны гипотония мышц, мышечная слабость (в том числе дыхательных и мимических мышц), аномалии развития скелета (сколиозы, удлиненная форма черепа, расщелины твердого нёба). Диагностика основывается на оценке статуса больного, изучении биоптата мышечной ткани, молекулярно-генетических исследованиях. Специфического лечения не существует, используется поддерживающая и симптоматическая терапия.
Общие сведения
Немалиновые миопатии (NEM) - совокупность генетических патологий со сходными клиническими проявлениями, которые характеризуются выраженным нарушением структуры мышечной ткани с формированием в ней аномальных нитеподобных или палочковидных структур. Впервые заболевание было описано в 1963-м году двумя независимыми группами исследователей под началом Д. Шая и П. Конена, в последующие годы было выявлено множество генетических разновидностей данного состояния. В зависимости от гена и мутации в нем, немалиновая миопатия может иметь как аутосомно-доминантный, так и аутосомно-рецессивный характер наследования. Встречаемость патологии на сегодняшний день не определена, но, по мнению многих врачей-генетиков, она является одной из наиболее распространенных врожденных миопатий - иногда указываются цифры 1:50 000. При этом достоверно определить этиологию немалиновой миопатии (ответственный за ее развитие ген и характер его дефекта) удается только в половине случаев, что указывает на незавершенность изучения данного заболевания.
Причины и классификация немалиновой миопатии
Причиной развития немалиновой миопатии являются генетические дефекты, приводящие к формированию аномальных форм структурных белков мышечной ткани - в основном, саркомерных протеинов. В результате этого нарушаются процессы сокращения мышц, а также, в ряде случаев, и их формирование, что приводит к появлению врожденных форм данного состояния. На сегодняшний день удалось идентифицировать семь генов, мутации которых ответственны за развитие немалиновой миопатии, однако, как было указано выше, они обуславливают лишь половину всех случаев заболевания. Кроме того, существует форма патологии с отсроченным началом, поражающая в основном взрослых людей - но, как показали более подробные исследования, она обусловлена приобретенными аутоиммунными нарушениями, а не генетическими факторами.
Помимо классификации, созданной на основе данных современной генетики, немалиновая миопатия также разделяется по своим фенотипическим проявлениям на врожденную (типичную, промежуточную и тяжелую разновидности) и ювенильную формы. Взаимосвязь этих двух систем классификации достаточно условна - различные мутации одного и того же гена могут приводить к развитию тяжелых и типичных врожденных форм или ювенильной разновидности заболевания.
NEM-1 - форма немалиновой миопатии, обусловленная мутацией гена TPM3, расположенного на 1-й хромосоме, регистрируется примерно в 2-3% случаев патологии. Ген кодирует одну из цепей медленного альфа-тропомиозина - структурного белка мышечной ткани. К немалиновой миопатии могут приводить различные мутации данного гена с разным типом наследования. Аутосомно-доминантные дефекты становятся причиной развития ювенильной формы заболевания, а аутосомно-рецессивные характеризуются врожденным тяжелым течением патологии.
NEM-2 - наиболее распространенный вариант немалиновой миопатии, составляющий примерно половину от всех генетически идентифицированных случаев данного заболевания. Вызывается дефектом гена NEB, который локализован на 2-й хромосоме и кодирует гигантский белок небулин. Последний функционально связан с тонкими (актиновыми) нитями мышечного волокна. Мутации гена NEB приводят к немалиновой миопатии исключительно врожденного характера с типичным или тяжелым течением.
NEM-3 - вторая по распространенности форма немалиновой миопатии, регистрируемая в 20-25% случаев заболевания с определенной этиологией. Причиной данной формы становятся мутации гена ACTA1, расположенного на 1-й хромосоме. Продуктом его экспрессии является альфа-актин скелетных мышц, один из важнейших структурных компонентов мышечной ткани. Выявлено более 180 типов мутаций данного гена, приводящих к самым разнообразным формам немалиновой миопатии с разным характером наследования, в основном - с врожденными нарушениями. Часть генетических дефектов ACTA1 являются спонтанными мутациями, возникающими de novo, по этой причине иногда возможна зародышевая мозаичность.
NEM-4 - разновидность немалиновой миопатии, составляющая примерно 3-4% всех случаев данного заболевания. Обусловлена дефектом гена TPM2, локализованного на 9-й хромосоме и кодирующего белок бета-тропомиозин, входящий в группу саркомерных протеинов. Мутации этого гена чаще всего передаются по аутосомно-доминантному механизму, имеются указания на возможность спонтанного повреждения гена. Этот генетический дефект проявляется врожденной типичной разновидностью немалиновой миопатии.
NEM-5 - форма немалиновой миопатии, диагностируемая исключительно в закрытых религиозных общинах амиш, где встречаемость этого заболевания достигает 1:500. Вызывается мутацией гена TNNT1, который располагается на 19-й хромосоме и отвечает за белок под названием медленный тропонин Т. Такие генетические нарушения приводят к тяжелой врожденной форме немалиновой миопатии с аутосомно-рецессивным механизмом наследования.
NEM-6 - редкая и малоизученная разновидность немалиновой миопатии, которая предположительно вызывается мутацией гена KBTBD13, локализованного на 15-й хромосоме. Достоверно неизвестно, какой именно белок кодирует данный ген, поэтому его роль в этиологии данного заболевания ставится под сомнение некоторыми исследователями. Предполагают, что его дефекты являются причиной аутосомно-доминантной немалиновой миопатии с легким течением и развитием в детском возрасте (ювенильная разновидность).
NEM-7 - также редкая форма немалиновой миопатии, которая была описана лишь у одной семьи в 2007-м году. Обусловлена дефектом гена CFL2, расположенного на 14-й хромосоме, продуктом его экспрессии является белок кофилин-2 (мышечный кофилин), принимающий активное участие в поддержании стабильного состояния актиновых нитей. Мутации CFL2 приводят к врожденной типичной форме заболевания с аутосомно-рецессивным характером наследования.
Симптомы немалиновой миопатии
Типичная (умеренная) разновидность является наиболее распространенной, сопровождается миотонией, затруднением при кормлении, антигравитационными движениями. Нарушения дыхания при этой форме немалиновой миопатии выражены незначительно, обычно сводятся к пониженной вентиляции легких по ночам. Слабость скелетных мышц в основном имеет проксимальный характер, прогрессирует крайне медленно, в ряде случаев больные способны передвигаться на костылях или ходить при помощи окружающих.
Промежуточная разновидность врожденной немалиновой миопатии представляет собой более тяжелую форму заболевания. Она характеризуется наличием всех вышеперечисленных нарушений, которые при этом имеют тенденцию к прогрессированию, возможны околосуставные ретракции, искривления позвоночника. Обычно к 6-7 годам больные этой формой немалиновой миопатии полностью утрачивают возможность передвигаться без инвалидного кресла, а к 10-12 годам - самостоятельно дышать, что приводит к летальному исходу. Тяжелая форма немалиновой миопатии проявляется резко выраженной миопатией в неонатальном периоде, затруднением глотания и сосания, в ряде случаев сильно страдает дыхательная мускулатура. Летальный исход наступает в первые месяцы жизни из-за дыхательной недостаточности или респираторной инфекции.
Диагностика и лечение немалиновой миопатии
Основными методами диагностики немалиновой миопатии являются изучение наследственного анамнеза пациента, гистологическое исследование мышечной ткани, электромиография и молекулярно-генетические исследования. В анамнезе больного могут определяться случаи заболевания у родственников, отсутствие таких примеров указывает на спорадический характер мутации. Патогистологические изменения в мышечной ткани сводятся к наличию в саркоплазме волокон нитевидных включений - при этом они могут иногда не определяться в мышцах младенцев даже при врожденных формах заболевания и появляться в более старшем возрасте. Иногда при немалиновой миопатии может обнаруживаться также неоднородность толщины мышечных волокон и их частичное разрушение. Электромиография при этом заболевании имеет признаки характерной «миопатической триады» - уменьшение амплитуды и длительности потенциалов действия в сочетании с их полифазностью. При помощи достижений современной генетики можно определить некоторые формы немалиновой миопатии - обусловленные мутациями генов NEB, ACTA1 и TPM2.
Специфического лечения немалиновой миопатии не существует, используют поддерживающую терапию, лечебную физкультуру, гимнастику для сохранения хотя бы минимальной активности мышц и предотвращения суставных контрактур. При нарушении дыхания применяют принудительную вентиляцию легких, питание младенцев в случае нарушений глотания или сосания осуществляют через назогастральный зонд. Если искривления позвоночника или другие скелетные нарушения могут усугубить состояние больного, прибегают к помощи хирургов-ортопедов.
Прогноз и профилактика немалиновой миопатии
В большинстве случаев врожденных разновидностей немалиновой миопатии прогноз заболевания неблагоприятный - у больных имеется высокий риск развития дыхательной недостаточности или респираторной инфекции, что может привести к летальному исходу. Лица с типичной формой патологии могут при адекватном уходе прожить до взрослого возраста, умственное и психическое развитие при этом генетическом заболевании не страдает. Намного более благоприятный прогноз у ювенильной немалиновой миопатии - жизненно важные группы мышц при этом типе патологии не поражаются. Тем не менее, подавляющее большинство больных рано или поздно оказываются прикованными к инвалидному креслу. Лицам с любой формой немалиновой миопатии очень важно следить за своей дыхательной системой и своевременно лечить любые инфекционные процессы в легких.
Миопатии
Миопатии — группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Причины миопатий
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр.), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и др.), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Патогенез
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и др. формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные - возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Симптомы миопатий
Большинство миопатий имеют постепенное начало с появления небольшой мышечной слабости в конечностях, более быстро возникающей усталости от ходьбы и другой физической нагрузки. В течение нескольких лет происходит нарастание слабости, появляются и прогрессируют мышечные атрофии, возникают деформации конечностей. Из-за значительной мышечной слабости пациенты с трудом поднимаются с пола и ходят по лестнице, не могут прыгать и бегать. Для того, чтобы встать со стула, им приходится использовать специальные приемы. Характерен вид больного: крыловидно отстоящие лопатки, опущенные плечи, выпяченный вперед живот и усиленный поясничный лордоз. Наблюдается «утиная походка» — пациент передвигается, раскачиваясь в стороны.
Патологические изменения при миопатиях происходят симметрично в мышцах конечностей и туловища. Как правило, мышечные атрофии наблюдаются в проксимальных отделах рук и ног. В связи с этим мышцы дистальных отделов конечностей могут выглядеть гипертрофированными. Такая миопатическая псевдогипертрофия наиболее заметна в мышцах голеней. Наряду с нарастанием мышечной слабости наблюдается постепенное угасание сухожильных рефлексов и прогрессирующее снижение мышечного тонуса, т. е. развивается и усугубляется периферический вялый паралич. Со временем результатом резкого ограничения активных движений становятся контрактуры суставов.
Миопатии могут сопровождаться поражением мимических мышц, что проявляется невозможностью вытянуть губы трубочкой, свистеть, нахмурить лоб или улыбнуться. Поражение круговой мышцы рта приводит к появлению дизартрии, связанной с затруднением произношения гласных звуков.
Клиника некоторых миопатий включает поражение дыхательной мускулатуры, приводящее к возникновению застойной пневмонии и развитию дыхательной недостаточности. Возможны патологические изменения сердечной мышцы с возникновением кардиомиопатии и сердечной недостаточности, мышц глотки и гортани с развитием дисфагии и миопатического пареза гортани.
Особенности отдельных форм миопатии
Ювенильная миопатия Эрба наследуется аутосомно-рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече-лопаточно-лицевая миопатия Ландузи - Дежерина имеет аутосомно-доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия — аутосомно-доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно-доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов - уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и др. ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение миопатий
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии и т. д.
Прогноз и профилактика
Наиболее неблагоприятны в прогностическом плане наследственные миопатии, проявляющиеся в раннем детском возрасте. В остальном прогноз зависит от формы миопатии, вовлеченности в процесс сердечной и дыхательных мышц. Прогноз вторичных миопатий более благоприятный при условии успешного лечения основного заболевания.
Профилактикой первичных миопатий служит тщательный сбор семейного анамнеза и обязательное консультирование у генетика пар, планирующих беременность. Профилактикой вторичных миопатий является исключение токсических воздействий на организм, своевременное лечение инфекционных и эндокринных заболеваний, коррекция метаболических нарушений.
Врожденная миопатия
Врожденная миопатия — врожденное заболевание, обусловленное генетически детерминированными нарушениями в строении мышечной ткани. Врожденная миопатия проявляется диффузной мышечной слабостью и снижением мышечного тонуса, выраженность которых значительно варьирует в зависимости от вида миопатии. В тяжелых случаях врожденная миопатия может привести к гибели ребенка от дыхательной недостаточности. Диагностируется врожденная миопатия в основном по результатам морфологического исследования образцов, полученных при биопсии мышц; электромиография, эргометрия и исследование мышечного тонуса имеют лишь вспомогательное значение. Врожденная миопатия может потребовать мероприятий по борьбе с дыхательными нарушениями, обеспечению зондового питания, коррекции имеющихся ортопедических деформаций и пр.
МКБ-10

Термин «врожденная миопатия» применяется в неврологии в отношении целой группы достаточно редких наследственных болезней, которые имеют сходную клиническую картину и дифференцируются лишь по специфическим морфологическим изменениям в строении мышечной ткани. К наиболее часто встречаемым видам врожденной миопатии относятся: болезнь центрального стержня, немалиновая миопатия, миотубулярная миопатия, миопатия с множественными стержнями и врожденная диспропорциональность типов мышечных волокон.
Врожденная миопатия является генетически обусловленным заболеванием. В зависимости от вида миопатии аномалия может локализоваться в различных локусах хромосом и передаваться по наследству доминантно, рецессивно или сцеплено с Х-хромосомой. Наличие аномального гена приводит к нарушению синтеза того или иного белка, входящего в структуру мышечной ткани. В результате изменяется строение мышечных волокон, что негативно отражается на их сократительной способности и приводит к генерализованной мышечной слабости. Обычно врожденная миопатия проявляется в раннем детском возрасте. Ее симптомы сохраняются в течение всей жизни пациента. В большинстве случаев врожденная миопатия характеризуется доброкачественным течением со слабым прогрессированием или вовсе без него.
Классификация врожденной миопатии
В основу классификации врожденной миопатии были положены 2 признака: наличие информации о локализации генной аномалии и данных о том, какой именно белок мышечной ткани является дефектным. В соответствии с этим выделяется врожденная миопатия с известным мутантным геном и определенным дефектным белком (немалиновая миопатия, болезнь центрального стержня, миотубулярная миопатия), врожденная миопатия с неопределенным дефектным белком, но установленным мутантным геном (десмин-связанная и актинзависимая миопатии) и врожденная миопатия, для которой неизвестными остаются и ген, и дефектный белок (врожденная диспропорция типов волокон, центронуклеарная миопатия, миопатия с множественными стержнями).
Симптомы врожденной миопатии
Врожденная миопатия в первые месяцы жизни ребенка характеризуется наличием синдрома «вялого ребенка»: диффузным снижением мышечного тонуса, легкой мышечной слабостью, плохим развитием мускулатуры и ослабленным сосанием. По мере развития ребенка мышечная слабость становится более заметной. Она проявляется невозможностью подняться с пола, залезть на стул, затруднениями при ходьбе и других действиях, которые без проблем выполняют другие дети того же возраста. Мышечная слабость при врожденной миопатии может быть выражена в различной степени. Обычно не наблюдается ее существенное прогрессирование. В тяжелых случаях ребенок так и не может встать на ноги и вынужден всю жизнь передвигаться на каталке. Но те навыки, которые были им приобретены, уже не утрачиваются.
Наибольшая опасность врожденной миопатии связана со слабостью дыхательной мускулатуры. При умеренной мышечной слабости отмечается постепенное развитие дыхательной недостаточности, частые бронхо-легочные заболевания (бронхит, очаговая пневмония, застойная пневмония и др.). Выраженная слабость дыхательных мышц может приводить к быстрому развитию дыхательной недостаточности и гибели ребенка в младенческом возрасте.
В некоторых случаях врожденная миопатия сочетается с дисморфичными чертами (высокое небо, удлиненная и узкая форма лица) и скелетными аномалиями (кифоз, сколиоз, косолапость, грудь сапожника, врожденный вывих бедра).
Характеристика отдельных видов врожденной миопатии
Болезнь центрального стержня наследуется аутосомно-доминантно, известны также отдельные спорадические случаи заболевания. Врожденная миопатия этого вида проявляется задержкой двигательного развития в период первого года жизни, реже обнаруживается у взрослых пациентов, часто сопровождается слабостью мимической мускулатуры. Характерен небольшой рост больных и хрупкая фигура, наличие скелетных деформаций. У пациентов с этим видом врожденной миопатии отмечается повышенный риск возникновения злокачественной гипертермии. В биоптате мышечной ткани выявляют мышечные волокна с единичными или множественными зонами асептического некроза.
Немалиновая врожденная миопатия включает актинопатию, небулинопатию, тропомиозинопатию и тропонинопатию. Ее наследование происходит чаще по аутосомно-доминантному принципу, но также встречается рецессивное наследование и спорадические случаи заболеваемости. Классическая форма немалиновой врожденной миопатии характеризуется синдромом вялого ребенка. Тяжелая форма проявляется еще во внутриутробном периоде в виде акинезии плода, а при рождении ребенка — тяжелыми двигательными нарушениями, слабостью мышц лица и дыхательной недостаточностью. Легкая форма этого типа врожденной миопатии диагностируется после периода раннего детства, иногда — в подростковом возрасте, и протекает без слабости лицевой мускулатуры. Существует также специфическая форма немалиновой врожденной миопатии, при которой возможно развитие офтальмоплегии, кардиомиопатии, синдрома ригидного позвоночника. Морфологическое исследование обнаруживает наличие в мышцах характерных палочко- или нитеподобных телец.
Миопатия с множественными стержнями чаще наблюдается как аутосомно-рецессивное заболевание, хотя возможен и доминантный тип наследования. Типична мышечная слабость в проксимальных отделах, которая наблюдается в грудном возрасте. Намного реже заболевание дебютирует в более старшем возрасте. В таких случаях отмечается генерализованная мышечная слабость. В мышечном биоптате определяются клетки с отсутствием митохондрий, деструкция сакромеров и гипотрофия мышечных волокон.
Врожденная диспропорция типов мышечных волокон проявляется генерализованной слабостью мышц, в том числе и лицевых, мышечной гипотонией, аномалиями скелета. Тип наследования этой врожденной миопатии пока не установлен. В биоптате мышц наблюдается увеличение количества и малый размер волокон I типа на фоне гипертрофии или нормального размера волокон II типа.
Диагностика врожденной миопатии
В легких случаях только прицельный осмотр с исследованием мышечной силы и тонуса позволяет неврологу заподозрить наличие у ребенка врожденной миопатии. Тщательное исследование мышц с проведением силового тестирования (эргометрии), стандартной и стимуляционной электромиографии, миотонометрии или электротонометрии позволяет получить дополнительные данные, свидетельствующие в пользу диагноза «врожденная миопатия». Исключить генерализованную воспалительную миопатию (дерматомиозит, полимиозит) и диффузный миозит позволяет отсутствие болевого синдрома, уплотнений и воспалительной отечности мышц.
Окончательно врожденная миопатия может быть диагностирована только после результатов морфологического исследования мышечной ткани, полученной путем биопсии мышц. Лишь это обследование позволяет определить специфичные для каждого вида миопатии изменения и установить точный диагноз. Однако даже биопсия не всегда позволяет достоверно верифицировать тип врожденной миопатии.
Лечение врожденной миопатии
К сожалению, на сегодняшний день не существует достаточно эффективных способов лечения и врожденная миопатия сохраняет свои проявления в течение всей жизни больного. Возможные методы терапии направлены на поддержание как можно более высокого уровня жизнеспособности пациента. Если врожденная миопатия сопровождается значительным снижением мышечной силы, в первые месяцы жизни медицинские мероприятия заключаются в борьбе с дыхательной недостаточностью, обеспечении питания через желудочный зонд, купировании бронхо-легочных осложнений. Благоприятное влияние на состояние больных в любом возрасте оказывает массаж, водолечение и физиотерапевтические процедуры. В более старшем возрасте может потребоваться ортопедическая коррекция имеющихся нарушений и социальная адаптация пациентов.
Врожденные миопатии - группа заболеваний, проявляющихся с рождения или в раннем детском возрасте снижением силы мышц, их гипотонией, ослаблением или отсутствием сухожильных рефлексов. Отличаются от прогрессирующих мышечных дистрофий доброкачественным течением. В ряде случаев симптомы дефицита мышечной системы остаются стабильными на протяжении всей жизни, иногда незначительно прогрессируют, возможно, даже регрессивное течение.
По клинической картине распознать эти заболевания чрезвычайно трудно, почти у всех больных имеется мышечная слабость, которая может быть генерализованной или преобладать в проксимальных отделах конечностей и мышцах плечевого и тазового пояса, имеются диффузная мышечная гипотрофия и гипотония, сухожильные рефлексы резко снижены или отсутствуют, иногда развиваются контрактуры.
При выраженной степени расстройств обычно ставится диагноз «floppy baby» (вялый ребенок).
Врожденная мышечная дистрофия - мышечные заболевания, имеющиеся с рождения или возникающие вскоре после рождения. Диагностика этих заболеваний может быть основана на следующих критериях:
- гипотония, слабость или артрогрипоз представлены с рождения;
- мышечная биопсия обнаруживает признаки миопатии (изменение размеров волокон, дегенерация волокон среднего размера, замещение мышечных волокон жировой тканью и коллагеном) и исключает денервацию;
- другие формы миопатии со специфическими клиническими признаками у новорожденных исключены.
Протокол "Врожденная миопатия"
Код по МКБ-10: G71.2
Врожденная мышечная дистрофия:
- со специфическими морфологическими поражениями мышечного волокна.
Диспропорция типов волокон
- немалиновая (болезнь немалинового тела).

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
1. Врожденные миопатии.
2. Болезнь центрального стержня.
3. Врожденная миопатия с диспропорцией типов волокон.
4. Миотубулярная (центронуклеарная) миопатия.
5. Острая миотубулярная миопатия.
6. Хроническая миотубулярная миопатия.
7. Немалиновая миопатия.
Диагностические критерии
Жалобы и анамнез: заболевание проявляется с рождения или в раннем детском возрасте; задержка в моторном развитии, гипотрофия, мышечная слабость, снижение тонуса и мышечной силы, нарушение походки.
Физикальные обследования
Болезнь центрального стержня
Наследуется по аутосомно-доминантному типу, характеризуется специфическими морфологическими изменениями, при которых в мышечных волокнах в центральной части имеются участки, заполненные компактными миофибриллами - "стержни". Диаметр волокон сохраняется, иногда он даже несколько увеличен.
Клиническая картина характеризуется более или менее значительной мышечной слабостью, преобладающей в проксимальных отделах конечностей, особенно нижних, и диффузной мышечной гипотонией.
Типично отставание в двигательном развитии при нормальном интеллекте. Мышечные атрофии выражены умеренно, сухожильные рефлексы сохранены или нерезко снижены.
Заболевание, как правило, не прогрессирует, к пубертатному периоду возможен регресс симптомов. Продолжительность жизни не ограничивается. Существуют субклинические формы.
Болезнь центрального ядра (миотубулярная миопатия)
Типы наследования: аутосомно-доминантный, аутосомно-рециссивный, рециссивный, сцепленный с Х-хромосомой. Эти обстоятельства говорят о генетической гетерогенности миотубулярной миопатии. Морфологические изменения при миотубулярной миопатии характеризуются уменьшением диаметра мышечных волокон. В центре этих малых волокон миофибриллы отсутствуют или очень рыхло упакованы.
Гистохимические реакции выявляют в центральных областях высокую активность окислительных ферментов. Измененные мышечные волокна в центре очень напоминают эмбриональные мышечные клетки или миотубулы. В связи с этим заболевание получило название митубулярной миопатии. Другое ее название - центронуклеарная, что обусловлено скоплением в центральной части мышечного волокна группы ядер.
Клиника характеризуется генерализованной мышечной гипотонией, слабостью глазодвигательных мышц (двусторонний умеренный птоз, парез наружных мышц глаза), умеренными костными деформациями.
Течение заболевания часто стационарное, но может иметь место регресс симптомов или со слабым прогрессированием. В большинстве случаев клинические симптомы отмечаются с первых дней жизни, но иногда проявляются впервые у взрослых.
Миопатия с врожденной диспропорцией типов волокон
Мышечные волокна делятся на два типа: I тип - красные волокна, II тип - белые. В последнее время II тип подразделяют на II A и II Б типы. В норме типы I и II имеют одинаковые размеры, их соотношение неодинаково в различных мышечных группах, но это различие довольно постоянно. При изучении врожденных форм миопатий с клинической картиной "floppy baby", было показано, что у некоторых больных патоморфологическое исследование выявляет единственный дефект - уменьшение в размерах типа I мышечных волокон и их преобладание, и сохранность величины увеличения в размере типа II волокон.
Иногда отмечается гипертрофия II A типа и отсутствие II Б типа волокон. Каких-либо других специфических изменений не выявляется. Клиническая картина характеризуется генерализованной слабостью, гипотонией, включающей дыхательную мускулатуру. В половине случаев отмечаются мышечные контрактуры разной степени выраженности.
Термин "врожденная миопатия с диспропорцией типов волокон" используется для описания новорожденных с гипотонией и иногда с артрогрипозом.
Немалиновая (нитеобразная) миопатия - наиболее частая форма. Патология состоит в наличии нитеобразных структур вдоль всего длинника пораженных мышечных клеток. Заболевание передается как по аутосомно-рециссивному, так и по аутосомно-доминантному типу. В основном семейные случаи заболевания, но все чаще спорадические случаи.
Клиника проявляется мышечной слабостью диффузного типа, включая мимические мышцы, которая отмечается с рождения. Слабость может распространяться на дыхательную мускулатуру.
Тяжесть состояния широко варьирует - от очень легкой до выраженной слабости с гипотрофией, приводящей к инвалидности. Больные отстают в двигательном развитии, позже начинают сидеть, ходить. Характерно сочетание мышечных симптомов с диспластическими изменениями скелета в виде вытянутой формы лицевого черепа, ретрогнатии «готического неба», деформации грудной клетки, позвоночника.
При биохимическом исследовании находят некоторое повышение активности креатинфосфокиназы в сыворотке крови.
Митохондриальные миопатии - группа миопатий, в которых специфическим и единственным морфологическим нарушением является патология митохондрий. Наследуются по аутосомно-рециссивному типу. Эта группа миопатий является гетерогенной, поскольку она представлена различными типами нарушения митохондрий.
При митохондриальных миопатиях морфологически может иметь место увеличение числа митохондрий (плеокониальная миопатия), увеличение их размеров (мегакониальная миопатия), неправильное расположение крист, наличие различных включений.
Плеокониальная миопатия - характеризуется эпизодическими приступами, начинающимися с жажды, рвоты, выраженной потребности в соли, профузного пота. Развивается мышечная слабость вплоть до вялой тетраплегии с отсутствием сухожильных рефлексов. Нарушается также речь и глотание. Приступ длится 10-14 дней. Выраженная мышечная слабость наблюдается с рождения, с возрастом может даже несколько уменьшаться. При морфологическом исследовании в мышечных волокнах обнаружены митохондрии гигантских размеров.
Илеокониальная миопатия - характеризуется медленно прогрессирующей слабостью и похуданием мышц проксимальных отделов конечностей. Отличительной особенностью заболевания явилось наличие приступов усиления мышечной слабости до состояния параличей. Такие приступы длились каждый раз от 10 до 14 дней, при этом появлялась потребность в приеме большого количества поваренной соли. При морфологическом исследовании биоптата мышц было обнаружено огромное количество митохондрий средней величины. Скопления митохондрий замещали миофибриллярный аппарат и располагались в околоядерных областях.
Врожденная мышечная дистрофия - гипотония и артрогрипоз определяются с рождения. Сухожильные рефлексы либо сохранны, либо отсутствуют, но из-за контрактур суставов их бывает трудно оценить. Врожденные контрактуры могут вовлекать различные суставы, но наиболее часто они приводят к кривошее и косолапости. Характерный симптом - врожденный вывих бедра.
Лабораторные исследования: возможно повышение уровня КФК в сыворотке крови.
Инструментальные исследования:
1. Компьютерная томография головного мозга для исключения органического поражения.
2. Электроэнцефалография (ЭЭГ) - изменения ЭЭГ неспецифичны.
3. Электромиография (ЭМГ) - выявляет первичный мышечный дефект, снижение амплитуды ЭМГ максимального напряжения, длительность потенциалов ДЕ уменьшена, увеличение числа полифазных потенциалов без увеличения их длительности, иногда ЭМГ имеет смешанный тип.
4. Магниторезонансная томография (МРТ) головного мозга - в настоящее время является методом выбора при диагностике пороков и аномалий и дегенеративных изменений головного мозга.
5. Нейросонография - выявляет наличие структурных изменений, расширение желудочковой системы, пороки и дизгенезии до закрытия родничка.
6. ЭКГ - изменения, указывающие на обменно-дистрофические изменения в миокарде.
Показания для консультаций специалистов:
1. Логопед - для выявления нарушения речи, дизартрий.
2. Психолог - для определения психического статуса ребенка.
3. Ортопед - для выявления контрактур, костных деформаций.
4. Протезист - для подбора ортопедической обуви, фиксирующих лонгет.
5. Окулист - осмотр глазного дна, выявление страбизма, нарушений зрения.
6. Кардиолог - выявление обменно-дистрофических и диспластических изменений сердца.
7. ЛОР-сурдолог - для определения тугоухости.
Минимум обследования при направлении в стационар:
1. Общий анализ крови.
2. Общий анализ мочи.
3. Кал на яйца глист.
Основные диагностические мероприятия:
10. Электромиография (ЭМГ).
14. Определение креатинкиназы в сыворотке крови.
Перечень дополнительных диагностических мероприятий:
1. Компьютерная томография головного мозга.
5. МРТ головного мозга.
7. R-графия грудной клетки.
8. УЗИ органов брюшной полости.
10. ЛОР сурдолог.
14. Мышечная биопсия.
Дифференциальный диагноз
Заболевания
Начало заболевания
ЭМГ
Течение заболевания
С рождения или в раннем детском возрасте, проявляется снижением мышечной силы и гипотонией мышц.
Снижение амплитуды ЭМГ максимального напряжения, длительность потенциалов ДЕ уменьшена, увеличение числа полифазных потенциалов.
Доброкачественное течение, стационарное или медленно прогрессирующее. При некоторых формах двигательные функции с возрастом улучшаются.
ДЦП. Атонически-астатическая форма
С рождения, на первый план выступает выраженная мышечная гипотония.
При произвольном сокращении регистрируется интерференционная кривая со сниженной амплитудой; фибрилляции, фасцикуляции отсутствуют.
Не прогрессирует, по мере роста и развития ребенка происходит регресс некоторых симптомов.
Заболевание начинается на 1-ом году жизни или обнаруживается уже при рождении. Спонтанная двигательная активность резко ослаблена.
Дегенерация мотонейронов спинного мозга, проявляющаяся ритмом частокола, II тип. В тяжелых случаях полное биоэлектрическое молчание.
Неуклонно прогрессирующее заболевание, обусловленное уменьшением и дегенерацией клеток мотонейронов спинного мозга.
Лечение
Тактика лечения
Цель лечения: улучшение трофики мышц и проведение импульса по нервному волокну и через мионевральный синапс; улучшение двигательной активности, социальная адаптация, профилактика патологических поз и деформаций.
Немедикаментозное лечение:
- массаж общий, тонизирующий;
- занятия с логопедом, психологом.
Медикаментозное лечение:
1. Нейропротекторы: церебролизин, актовегин, пирацетам, пиритинол, гинкго-билоба.
2. Стимулирующая терапия: прозерин, дибазол, галантамин, оксазил, нейромидин.
3. Ангиопротекторы: винпоцетин, циннаризин.
4. Витаминотерапия: витамины группы В - тиамин бромид, пиридоксин гидрохлорид, цианокобаламин, аевит, фолиевая кислота, токоферол, ретинол, эргокальциферрол.
5. Комплексы аминокислот: метионин, глицин.
6. Анаболические стероиды: неробол, ретаболил, оротат калия.
Профилактика осложнений:
- предупреждение контрактур, патологических поз;
- профилактика вирусных и бактериальных инфекций.
Дальнейшее ведение:
- регулярные занятия ЛФК;
- обучение родителей навыкам массажа, ЛФК;
- носить ортопедическую обувь.
Перечень основных медикаментов:
1. Аевит, капсулы
2. Актовегин ампулы по 80 мг, 2 мл
3. Нейромультивит (витамины В1, В6, В12), таблетки
4. Оротат калия, таблетки
5. Пирацетам, ампулы по 5 мл, 20%
6. Пиридоксин гидрохлорид, ампулы по 1 мл, 5%
7. Прозерин, ампулы 0,05%, 1 мл
8. Прозерин, таблетки 0,015
9. Тиамин хлорид ампулы, 1 мл, 5%
10. Фолиевая кислота, таблетки 0,001
11. Цианокобаламин, ампулы 200 и 500 мкг
Дополнительные медикаменты:
1. Винпоцетин, таблетки 5 мг
2. Галантамин, ампулы 0,25%, 1мл
3. Гинкго-Билоба, (танакан) таблетки 40 мг
4. Глицин, таблетки 0,1
5. Дибазол, таблетки 0,02
6. Колекальциферол 1 мл, 15 тыс. МЕ
7. Луцетам, таблетки 0,4
8. Магне В6, таблетки
9. Нандролона деканоат (ретаболил), ампулы 1 мл, 50 мг
10. Неуробекс, таблетки
11. Оксазил, таблетки 0,001
12. Пирацетам, таблетки 0,2
13. Пиритинол, суспензия или таблетки 0,1
14. Токоферолола ацетат (витамин Е), капсулы 200 МЕ
15. Церебролизин, ампулы 1 мл
16. Циннаризин, таблетки 25 мг
Индикаторы эффективности лечения:
1. Улучшение двигательной активности.
2. Улучшение мышечного тонуса.
3. Повышение силы мышц.
4. Приобретение навыков самообслуживания.
5. Улучшение эмоционального и психического тонуса ребенка.
Госпитализация
Показания к госпитализации (плановая): двигательные расстройства, мышечная слабость, ослабление или отсутствие сухожильных рефлексов, нарушение движений, походки, костные деформации.
Читайте также:
- Розовый лишай. Причины, диагностика и лечение
- Анатомия: Лимфатические узлы и сосуды головы. Топография, строение, расположение лимфатических узлов и сосудов головы.
- Перелом диафиза лучевой кости. Диагностика и лечение
- Показания для восстановления дистального сухожилия двуглавой мышцы плеча
- Лечение медиастинита