
Модели вирусов и бактерий
![]()
![]()
![]()
Рейтинг топ-10 по версии КП
![]()
Качественный респиратор от вирусов, которых состоит из шести слоев. Защитит дыхательные органы не только от инфекций, но от пыли и взвешенных частиц. Оборудован угольным фильтром. От болезней он, конечно, не защищает. Эта часть больше для обеззараживания токсичных аэрозольных частиц. Один из слоев респиратора - мелтбаун. Это сравнительно новый материал, который используют в медицинских масках. У него высокие барьерные свойства - то есть по максимум тормозит проникновение опасных частиц извне в дыхательную систему. Производитель также хвастает, что при изготовлении была использована ультразвуковая сварка. То есть составляющие маски скреплялись не клеем или путем нагревания, а посредством мощного ультразвука. Технология, к слову, не такая уж новая, но в легкой промышленности все же редкая.
+ Большая коробка, внутри которой респираторы в индивидуальной упаковке
+ Шесть защитных слоев
Основные характеристики
Вид маски - процедурная, количество в упаковке - 25 шт., шестислойная, нестерильная, одноразовая.
![]()
![]()
Этот респиратор гарантирует максимальную степень защиты. Мощнее будет только устройства со сменными фильтрами, но такие не станешь носить в обычной жизни из-за массивности. Эта модель респиратора отвечает классу защиты FFP3. Согласно характеристикам, он обеспечивают фильтрацию 99% твердых и жидких частиц. Конструкция такова, что маска надежно прилегает к лицу. Причем это прочная модель, ее сложно измять, а значит повредить ее защитные свойства. Производитель рассказывает, что для использовал высокотехнологичный фильтрующий материал. При всем этом, изделие встречает низкое сопротивление дыханию, что сделано для комфорта носящего. На лицевой части есть клапан выдоха. Он устроен таким образом, что воздух устремляется вниз. Это помогает отводить влагу. Плюс те, кто носят очки, оценят, что стекла не будут запотевать. Для этого же, кстати, выполнена особенная форма верхней части - для лучшей совместимости с линзами.
+ Высокий уровень защиты
+ Качество
Основные характеристики
Тип - респиратор-полумаска, количество в упаковке - 1 шт., нестерильная, одноразовая.
![]()
Еще один респиратор высокого класса защиты от вирусов и не только. Строго говоря, его разрабатывали для использования на производствах, чтобы снизить воздействие вредных веществ на дыхательные органы рабочих. Но и функцию защиты от вирусов он выполнит. Маска оборудована уже знакомым нам клапаном. Отверстие отводит выдыхаемый воздух из маски, чтобы внутри не скапливались тепло и влага - благоприятная среда для бактерий. Обратите внимание и на форму изделия. Она не громоздкая, что удобно. Класс защиты, как и у предыдущей модели FFP3. Отдельно отметим гибкую системы настройки ремешком. Очень часто, респираторы от вирусов не оборудованы подобной системой и сильно давят на голову.
+ Регулируемые тесемки
+ Форма
Основные характеристики
Тип - респиратор-полумаска, количество в упаковке - 1 шт., нестерильная, одноразовая.
В нашем первом посте про трехмерное моделирование вирусов мы перечислили основные стадии процесса и рассказали о том, с чего мы начинаем и как собираем исходную информацию. В этой заметке мы расскажем о следующем этапе работы — о создании моделей отдельных молекул, из которых впоследствии будет собрана целая частица.

Компоненты вирусной частицы Гриппа A/H1N1
Вирусная частица — это молекулярный механизм, решающий две принципиальные задачи. Во-первых, частица должна обеспечить упаковку вирусного генома и его защиту от деструктивных факторов среды, пока вирус путешествует из клетки, в которой он собрался, к клетке, которую он сможет заразить. Во-вторых, частица должна быть способна присоединиться к заражаемой клетке, после чего доставить вирусный геном и сопутствующие молекулы внутрь, чтобы запустить новый цикл размножения. Задач не очень много, поэтому вирусы, за редким исключением, могут позволить себе быть довольно экономными в том, что касается структуры.
В частности, геном большинства вирусов невелик и кодирует не очень много белков, нередко это число меньше 10. При этом вирус может заставить клетку синтезировать большое количество однотипных белков, из которых потом соберется вирусная оболочка — капсид. Таким образом, вирусные частицы обычно состоят из большого числа одинаковых элементов, которые связываются друг с другом как детали конструктора, часто образуя регулярные и симметричные структуры. Так, очень многие, хоть и не все вирусные упаковки или их фрагменты имеют спиральную или икосаэдрическую форму.

Примеры вирусных капсидов с икосаэдрической симметрией. Молекула бактриородопсина в правом нижнем углу — для сравнения. (Иллюстрация из обзора).
Для сборки модели вируса принципиально важно знать, как устроены отдельные белки общей структуры и как они друг с другом связываются, эту структуру формируя. Современная наука владеет целым набором методов, которые могут дать ответы на эти вопросы, однако ни один из подходов, к сожалению, не является универсальным и решает только часть задач которые стоят перед нами при создании научно достоверных моделей вирусов с атомной детализацией.
Напомним, что белки — это полимерные молекулы, состоящие из последоватльно связанных между собой мономеров — аминокислот. В водных растворах белки обычно сворачиваются в сложные трехмерные глобулы (почти как головоломка “Змейка Рубика”), форма которых зависит от аминокислотного состава и некоторых других факторов. Пространственное строение этих глобул определяют в основном методами рентгеноструктурного анализа и ЯМР-спектроскопии. Также в последнее время к этой задаче позволяет подойти электронная микроскопия.
В целом, методы определения пространственной структуры молекул сложны и имеют целый набор ограничений, поэтому далеко не все вирусные белки описаны полностью. Так, рентгеноструктурный анализ предполагает наличие кристалла, через который пропускается рентгеновское излучение. Атомы кристалла провоцируют дифракцию рентгеновских лучей, по картине которой можно оценить распределение электронных плотностей в кристалле, а по этим данным уже восстановить расположения конкретных атомов. Этот метод дает разрешение вплоть до чуть более 1 ангстрема (0,1 нм), однако в случае белков проблема заключается в том, что далеко не все из них можно кристаллизовать. Особенно сложным это оказывается, если белок имеет гибкие подвижные или заякоренные в мембране фрагменты.
ЯМР-спектроскопия основана на явлении ядерного магнитного резонанса и позволяет описывать строение белков в растворе. Этот подход выявляет набор возможных положений атомов в молекуле и, в отличие от предыдущего метода, дает возможность оценить степень гибкости тех или иных ее участков. Но ЯМР-спектроскопия хорошо работает только для сравнительно небольших молекул, поскольку крупные белки дают слишком много шума.
Электронная микроскопия позволяет описать строение крупных молекулярных комплексов, что бывает очень полезно, когда речь идет о вирусах. Для многих симметричных структур можно получить большой набор изображений под разными углами, проанализировав которые можно воссоздать трехмерную картину. Для отдельных объектов разрешение, получаемое в результате применения разных вариантов электронной микроскопии (до 4-5 ангстрем), оказывается не многим хуже разрешения рентгеноструктурного анализа, хотя обычно для получения полной информации приходится совмещать разные подходы и, например, “вписывать” структуры отдельных белков в карты электронных плотностей, получаемые при помощи электронной микроскопии.

Структуры тримера белка оболочки ВИЧ (красные и голубые фрагменты молекул) в комплексе с участком одного из антител к этому белку (зеленые и желтые фрагменты), вписанные в карту электронной плотности, полученную методом крио-электронной микроскопии с разрешением 9 ангстрем. Из статьи Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation.
Как мы писали в прошлом посте, получаемые структуры систематизируются и хранятся в базе данных Protein Data Bank. При этом в формате *.pdb записываются координаты атомов, и существует целый набор программ, позволяющих эти данные визуализировать и работать с такими структурами. Среди них, например VMD, Chimera, PyMol и десятки других.

Скриншот текстового отображания файла в формате *.pdb. Описываются координаты отдельных атомов в аминокислотах белка.
Программы могут отображать белки несколькими способами. Помимо простого отображения атомов сферами разного диаметра, соответствующего ван-дер-ваальсовым радиусам атомов, существует возможность показать отдельные связи, поверхность молекулы, а также изгибы аминокислотной цепочки при помощи структур, напоминающих ленты (ribbon diagram), которые наглядно демонстрируют, где в белке аминокислоты образуют альфа-спирали, где бета-слои, а где неструктурированные участки.

Различные варианты визуализации структуры наружней части гемагглютинина вируса гриппа в программе Chimera.
В качестве отступления, надо сказать, что программы, в которых обычно работают ученые, визуализируя отдельные молекулы или белковые комплексы, чаще всего позволяют получить лишь довольно примитивные с эстетической точки зрения результаты (достаточно, например, посмотреть на несколько скриншотов из программы VMD). Принципиально более широкие возможности открываются, если импортировать модели молекул в программы, которые используют профессиональные дизайнеры и специалисты компьютерной трехмерной графики. Эти программы в сочетании с плагинами, улучшающими качество рендера, позволяют получать действительно интересные и привлекательные визуализации. Мы еще расскажем об этом в следующих постах. Пока просто приведем пример:
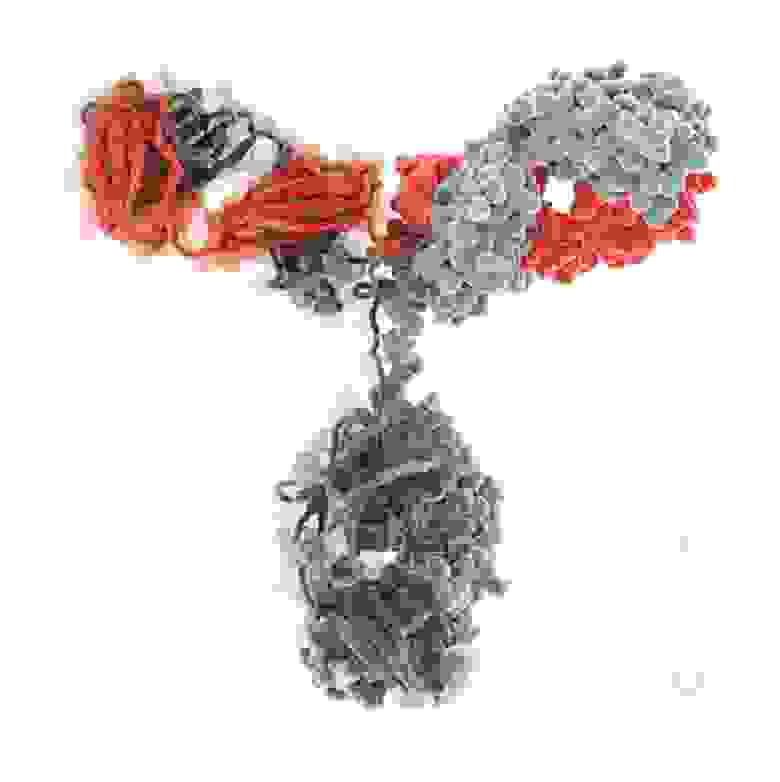

Изображения молекулы иммуноглобулина G.

Шаблоны для моделирования нейраминидазного комплекса вируса гриппа. А — фрагмент мономера нейраминидазы N2 из структуры 2AEP в базе данных PDB, B — “стебель” гемагглютинин-нейраминидазы парагриппа (3TSI), С — трансмембранный пептид 2LAT. D — финальная полученная модель.
Окончательная модель белка обычно создается с учетом известных структур его фрагментов, найденных разными методами шаблонов, а также моделей от сервера I-Tasser. Для этого используется программа Modeller. Она позволяет строить модель по гомологии с использованием одного или нескольких шаблонов, а также вносить дополнительные модификации, например, создавать дисульфидные связи в заданных местах.
Другим важным аспектом строения вирусов, информация о котором в научной литературе часто оказывается не полна, является взаимодействие между отдельными белками. В нашем случае от этого зависит то, какими поверхностями модели отдельных белков будут контактировать друг с другом и другими компонентами вириона в финальной модели. Информацию о взаимодействиях тоже позволяет уточнить структурная биоинформатика.
Программа докинга не моделирует естественный процесс образования комплекса, это было бы слишком медленно и ресурсоемко, а перебирает варианты взаимного положения двух или более молекул в поисках наилучшей структуры. При докинге обычно большую молекулу в комплексе называют рецептором, а меньшую — лигандом. Для определения качества структуры комплекса лиганда с рецептором используются различные оценочные функции. В идеале в качестве такой функции должна выступать свободная энергия системы, но она слишком сложно вычисляется, поэтому применяют различные эмпирические псевдопотенциалы, учитывающие потенциальную энергию (которая как раз вычисляется просто), площадь контакта лиганда и рецептора, соответствие различным правилам, которые исследователи вывели из анализа большого числа комплексов, и всякие загадочные слагаемые, не имеющие физического смысла, но улучшающие результат программы при испытании на большом количестве известных комплексов. Поиск минимума такого псевдопотенциала в современных программах обычно происходит с помощью различных вариаций метода Монте-Карло и генетических алгоритмов. В настоящее время существует множество программ молекулярного докинга (наиболее известные из них — Dock, Autodock, GOLD, Flexx, Glide), отличающиеся оценочными функциями, методами минимизации и дополнительными возможностями. При этом во время поиска молекулы рецептора и лиганда могут как оставаться неподвижными (такой тип докинга называется жестким), так и несколько менять конформацию (гибкий докинг). Очевидно, что второй вариант более ресурсоемкий, но и результаты такого поиска обычно правдоподобнее. Докинг малых молекул к белкам сейчас является стандартным этапом разработки новых лекарственных препаратов. Можно, например, провести докинги для 10 миллионов лигандов, и выбрать сотню наиболее перспективных соединений для дальнейшей экспериментальной работы — это называется виртуальный скрининг.
Помимо исследований небольших молекул, докинг может быть использован и для построения белок-белковых и белок-нуклеотидных комплексов. Для этих целей также разработано большое количество программ и онлайн-сервисов (ZDOCK, pyDOCK, HEX). Например, в ходе нашей работы над вирусом папилломы человека (ВПЧ) мы столкнулись с тем, что, несмотря на наличие полной структуры внешнего слоя капсида, образованнного белком L1, совершенно не было информации о строении белка L2, который в капсиде расположен ближе к геному, а соответственно, нет данных о том, как пентамеры L1 взаимодействуют с молекулами L2. Мы построили модель белка L2 по гомологии, используя сервер Tasser, после чего провели докинг в программе HeX. В ходе докинга роль рецептора выполнял пентамер L1. Именно на его поверхности проводился поиск оптимального места посадки L2. При этом все структуры оставались неподвижными. Т.е. использовался метод жесткого докинга. В результате была получена правдоподобная структура комплекса пентамера, собранного из L1 и минорного белка L2.
Наконец, биоинформатическими методами можно пытаться восстановить то, какие изменения в структуру вирусных белков вносит сама клетка, в которой они образуются. Большинство белков после синтеза подвергаются дополнительным химическим посттрансляционным модификациям (ПТМ), которые могут серьезно влиять на выполняемые белком функции. Среди таких модификаций фосфорилирование, убиквитинирование, гликозилирование, нитрозилирование, внесние разрывов и другие химические изменения. Многие поверхностные белки вирусов гликозилированы, причем эта модификация имеет непосредственное значение для выполнения основной функции поверхностных белков вируса — связывания с клеточными рецепторами. С другой стороны, белки вирусных матриксов — слоев, которые встречаются непосредственно под липидными оболочками некоторых вирусов, для заякоривания в мембране часто должны быть связаны, например, с миристиловой кислотой — небольшой гидрофобной молекулой, облегчающей взаимодействие белков с липидами. Таким образом, в нашей работе модификации белков тоже требуют внимания.
В настоящее время возможные ПТМ достаточно сложно предсказываются. Основные существующие методы и сервисы основаны на поиске соответствующей экспериментальной информации для сходных белков или поиске в последовательности исследуемого белка небольших участков, характерных для того или иного типа модификации.
В нашей работе при подготовке моделей мы пользуемся экспериментальной информацией, отраженной в соответствующей записи базы данных UNIPROT.
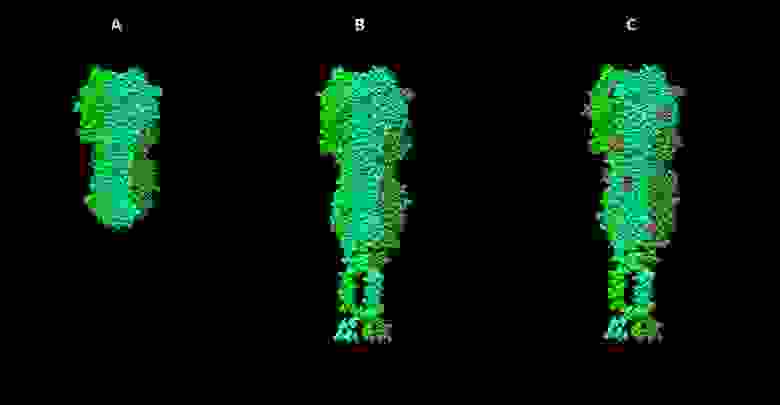
Стадии работы над моделью гемагглютинина вируса гриппа. А — визуализация структуры 3ZTJ из базы данных PDB. B — модель гемагглютинина вируса гриппа H1N1, построенная на основе гомологии с 3ZTJ с достраиванием трансмембранных участков молекулы. С — модель с учетом посттрансляционных модификаций (гликозилирования).
Последнее, о чем хочется упомянуть, — это то, что при подготовке новых моделей белков и, особенно, их комплексов, необходимо проводить оптимизацию структур. Наиболее простым методом оптимизации является минимизация энергии. Она используется для достаточно быстрого “спуска” системы в локальный минимум потенциальной энергии. Эту манипуляцию желательно проводить после каждой модификации структуры молекул. Она позволяет избежать таких неприятностей, как перекрывание атомов или появление неправильных длин связей. Различные методы минимизации энергии предусмотрены практически в любом программном пакете молекулярного моделирования.
Стоит отметить, что данный метод позволяет провести лишь предварительную и очень грубую оптимизацию. Для более точной подготовки пространственных структур используются методы молекулярной динамики или квантовой механики. Последние, например, используются для наилучшей оптимизации структуры небольших молекул лигандов и наиболее точных расчетов энергии межмолекулярных взаимодействий. Но, наибольшая точность, что вполне логично, связана с более ресурсоемкими вычислениями, что делает эти методы практически неподъемными в применении к большим биологическим макромолекулам.
Оценить поведение и стабильность структур достаточно массивных молекул, таких как полипептиды и нуклеиновые кислоты позволяют методы молекулярной динамики.
Метод молекулярной динамики заключается в изучении поведения атомов и молекул и их движений во времени. Расчеты молекулярной динамики позволяют, например, исследовать стабильность как отдельных молекул, так и их комплексов, позволяют оценить значимость возможных конформационных перестроек, влияние точечных мутаций и многое другое. Современные методы анализа результатов симуляций молекулярной динамики позволяют получить самые подробные сведения о поведении во времени как отдельных атомов, так и всей исследуемой системы.
В зависимости от того, насколько хорошо изучены белки того вируса, модель которого мы хотим создать, каждый раз приходится подбирать подходы для достройки и оптимизации моделей всех белков и их взаимодействий. После того, как все структуры получены, можно приступать к сборке полной модели. О том, как это делается, мы расскажем в следующих постах серии о создании научно достверных моделей вирусов человека.
PS:
Ставшая лидером в опросе прошлого поста тема Медицинская анатомическая иллюстрация — история изучения тела человека в работах иллюстраторов 5 столетий будет следующей. С потрясающими гравюрами, восковым моделями прошлого века, пластификатами трупов, атласами выдающся исследователей, 3Д реконструкциями на основе послойных срезов замороженного смертника, интерактивными приложениями и работами современных медицинских иллюстраторов. Скоро.
Открытие вирусов
В 1892 году Д.И. Ивановский (см. Рис. 1), изучая мозаичную болезнь табака (см. Рис. 2), установил, что причиной заболевания является некое инфекционное начало, содержащееся в листьях больных растений, которое проходит через фильтр, задерживающий обыкновенные бактерии. Если профильтрованный сок внести в листья здоровых растений, то они также заболевают мозаичной болезнью.

Рис. 1. Д.И. Ивановский

Рис. 2. Мозаичная болезнь табака
В 1898 году независимо от Ивановского аналогичные результаты получил голландский микробиолог М. Бейеринк. Однако он предположил, что мозаичную болезнь табака вызывают не мельчайшие бактерии, а некое жидкое заразное начало, которое он назвал фильтрующим вирусом.
Размеры вирусов определяются нанометрами (20-200 нм), поэтому их изучение началось после открытия электронного микроскопа. В настоящее время описаны вирусы практически всех групп живых организмов.
Строение вирусов
Вирусы – неклеточные формы жизни. Они состоят (см. Рис. 3) из фрагмента генетического материала (РНК или ДНК), составляющего сердцевину вируса, и защитной оболочки, которая называется капсид. У некоторых вирусов (герпес, грипп) есть дополнительная липопротеидная оболочка – суперкапсид, которая возникает из плазматической мембраны клетки-хозяина.

Рис. 3. Строение вируса
Вирусы не способны к самостоятельной жизнедеятельности. Они могут проявлять свойства живого, только попав в клетку-хозяина. Они используют потенциал и энергию этой клетки для создания своих новых вирусных частиц, следовательно, вирусы являются внутриклеточными паразитами.
Размножение вирусов
Обычно вирус связывается с поверхностью клетки-хозяина и проникает внутрь. Каждый вирус ищет своего хозяина, то есть клетки строго определенного вида. Например, вирус – возбудитель гепатита (желтуха) проникает и размножается только в клетках печени, а вирус эпидемического паротита (свинка) – только в клетках околоушных слюнных желез человека.
Проникнув внутрь клетки-хозяина, вирусная ДНК или РНК начинает взаимодействовать с ее генетическим аппаратом таким образом, что клетка начинает синтезировать белки, свойственные вирусу (см. Рис. 4).

Рис. 4. Схема репродукции вируса
При заражении ретровирусом (например, вирус иммунодефицита человека (ВИЧ)), у которого в качестве генетического материала используется молекула РНК, наблюдается другая картина. При попадании ретровируса в клетку-хозяина происходит обратная транскрипция. То есть на основе вирусной РНК синтезируется вирусная ДНК, которая встраивается в ДНК человека. Такой тип взаимодействия вируса с клеткой называется интегративным, а встроенная в состав хромосомы клетки ДНК вируса называется провирусом. Далее провирус реплицируется (удваивается) в составе хромосомы и переходит в геном дочерних клеток. Однако под влиянием некоторых физических и химических факторов провирус может выщепляться из хромосомы клетки и переходить к продуктивному типу взаимодействия, то есть синтезировать новые вирусные частицы.
При заражении ВИЧ человек чувствует себя здоровым, пока вирусный генетический материал встроен в хромосому человека. Однако при выщеплении этого вирусного генетического материала из клетки она начинает образовывать новые вирусные частицы, вследствие чего развивается смертельное заболевание – синдром приобретенного иммунодефицита (СПИД).
Вирусы являются возбудителями большого количества заболеваний человека: корь, грипп, оспа, краснуха, энцефалит, свинка, гепатиты, СПИД. Известен также целый ряд заболеваний растений, вызываемых вирусами, например мозаичная болезнь табака, томатов, огурцов или скручивание листьев картофеля. Всего описано около 500 видов вирусов, поражающих клетки позвоночных животных, и около 300 вирусов растений. Некоторые вирусы участвуют в злокачественном перерождении клеток и тем самым провоцируют онкологические заболевания.
ДНК- и РНК-содержащие вирусы
В зависимости от содержащегося генетического материала вирусы подразделяются на ДНК-содержащие и РНК-содержащие.
Одноцепочные РНК-содержащие вирусы подразделяются на:
1. Плюс-нитевые (положительные). Плюс-нить РНК этих вирусов выполняет наследственную (геномную) функцию и функцию информационной РНК (иРНК).
2. Минус-нитевые (отрицательные). Минус-нить РНК этих вирусов выполняет только наследственную функцию.
К РНК-содержащим вирусам относятся более 
вирусов, вызывающих респираторные заболевания, а также вирус гриппа, кори, краснухи, свинки, ВИЧ. Также существует специфическая группа вирусов – арбовирусы, которые переносятся членистоногими.
Двухцепочные ДНК-содержащие вирусы вызывают такие заболевания, как папиллома человека или герпес, гепатит В (гепатит А и гепатит С вызывается РНК-содержащими вирусами).
ДНК-содержащие вирусы поражают также растения. Они вызывают, например, золотую мозаику бобов или полосатость у кукурузы.
Вирус гепатита С
По своему строению вирус гепатита С – это РНК-содержащий вирус, имеющий сферическую форму, сложно устроенный (см. Рис. 5).
В качестве генетического материала такой вирус содержит линейную однонитчатую молекулу РНК.

Рис. 5. Гепатит С
Вопреки бытующим предрассудкам, подцепить вирус гепатита C невозможно через социальные контакты (поцелуи, объятия), через продукты или воду, через грудное молоко. Вы ничем не рискнете, если разделите с носителем вируса трапезу или напитки. Заразиться гепатитом C можно при контакте с кровью инфицированного человека либо половым путем.
В настоящее время для лечения гепатита С используют два препарата: Интерферон альфа и Рибавирин.
Бактериофаги

Рис. 6. Бактериофаг (Источник)
Особую группу вирусов составляют бактериофаги (или просто фаги), которые заражают бактериальные клетки (см. Рис. 6). Фаг укрепляется на поверхности бактерии при помощи специальных ножек и вводит в ее цитоплазму полый стержень, через который проталкивает внутрь клетки свою ДНК или РНК. Таким образом, генетический материал фага попадает внутрь бактериальной клетки, а капсид остается снаружи. В цитоплазме начинается репликация генетического материала фага, синтез его белков, построение капсида и сборка новых фагов. Уже через 10 мин после заражения в бактерии формируются новые фаги, а через полчаса бактериальная клетка разрушается, и из нее выходят около 200 заново сформированных вирусов – фагов, способных заражать другие бактериальные клетки (см. Рис. 7). Некоторые фаги используются человеком для борьбы с болезнетворными бактериями, вызывающими холеру, дизентерию, брюшной тиф.

Рис. 7. Схема размножения бактериофага (Источник)
Список литературы
- Каменский А.А., Криксунов Е.А., Пасечник В.В. Общая биология 10-11 класс Дрофа, 2005.
- Биология. 10 класс. Общая биология. Базовый уровень / П.В. Ижевский, О.А. Корнилова, Т.Е. Лощилина и др. – 2-е изд., переработанное. – Вентана-Граф, 2010. – 224 стр.
- Беляев Д.К. Биология 10-11 класс. Общая биология. Базовый уровень. – 11-е изд., стереотип. – М.: Просвещение, 2012. – 304 с.
- Агафонова И.Б., Захарова Е.Т., Сивоглазов В.И. Биология 10-11 класс. Общая биология. Базовый уровень. – 6-е изд., доп. – Дрофа, 2010. – 384 с.
Дополнительные рекомендованные ссылки на ресурсы сети Интернет
Домашнее задание
Если вы нашли ошибку или неработающую ссылку, пожалуйста, сообщите нам – сделайте свой вклад в развитие проекта. 

Читайте также: