
Дермато поли миозит при новообразованиях
Дерматомиозит и полимиозит – это заболевания, характеризующиеся системным негнойным воспалением соединительной ткани, поперечно-полосатой и гладкой мускулатуры с нарушением её двигательной функции.
- При дерматомиозите (болезнь Вагнера, болезнь Вагнера-Унферрихта-Хеппа) поражение мышц сочетается с поражением кожи в виде эритемы и отека на открытых участках тела.
- При полимиозите развивается только поражение мышц, а кожный синдром отсутствует.
Заболеваемость составляет примерно 1:200000 в год. Девочки и женщины болеют в 2 раза чаще.
Этиология и патогенез
Причина заболевания неизвестна. Существует мнение, что в его развитии имеют значение вирусная инфекция и аутоиммунные реакции. В крови больных дерматомиозитом детей обнаружены антитела к пикорнавирусам, а у больных полимиозитом взрослых к Toxoplazma gondii. У больных гриппом и инфекцией, вызванной вирусом Коксаки, может развиться легкий миозит. Но до настоящего времени вирусологические и серологические исследования и отсутствие эффекта от антибактериальной терапии токсоплазмоза не подтверждают вирусную природу этих заболеваний. Но, возможно, измененные вирусы содержат антигены, напоминающие собственные антигены организма и вызывающие образование аутоантител к цитоплазматическим белкам (антигену J0-1).
Аутоиммунная природа заболеваний подтверждается обнаружением аутоантител к ядерным или цитоплазматическим антигенам, культурам эпителиальных клеток человека (Нер-2), синтетазам транспортной РНК (например, к гистидил-тРНК-синтетазе) и внутрисосудистых отложений иммунных комплексов, повреждающих эндотелиальные клетки. Иммуногистохимические исследования мышечной ткани при полимиозите выявляют воспалительные инфильтраты, содержащие активированные цитотоксические лимфоциты СD8 наряду с лимфоцитами СD4 и макрофагами. При дерматомиозите в биоптатах мышц обнаруживают отложения иммуноглобулинов и компонентов комплемента С5b–С9, а вокруг мелких сосудов накапливаются СD4 + -клетки и β-лимфоциты. Развивается поражение сосудов, опосредованное антителами и приводящее к образованию очагов некроза в мышечных волокнах, их дегенерации и потере поперечной исчерченности.
Регенерация скелетных мышц проявляется увеличением числа мышечных ядер и базофилией мышечных элементов. В последующем развивается интерстициальный фиброз.
Классификация
В настоящее время используется классификация (по R.L. Woltman, 1994), в которую вошли идиопатические воспалительные миопатии и миопатии, вызываемые инфекциями, лекарствами и токсинами.
- Идиопатические воспалительные миопатии:
- Первичный полимиозит.
- Первичный дерматомиозит.
- Ювенильный дерматомиозит.
- Миозит, ассоциирующийся с опухолями.
- Миозит с "включениями".
- Миозит, ассоциирующийся с эозинофилией.
- Локализованный или очаговый миозит.
- Оссифицирующий миозит.
- Гигантоклеточный миозит.
- Миопатии, вызываемые инфекциями.
- Миопатии, вызываемые лекарственными средствами.
Симптомы
Первичный полимиозит. Может начинаться внезапно или постепенно. Вначале развивается симметричная мышечная слабость проксимальных мышц конечностей, особенно бедер. Больным трудно подниматься и спускаться по лестнице, вставать из положения на корточках. При поражении мышц плечевого пояса больные испытывают затруднение при подъеме рук вверх и причесывании. Нередко поражаются мышцы шеи, что сопровождается нарушением ее сгибания. У многих больных развивается слабость поперечнополосатых мышц глотки и верхней трети пищевода, приводящая к дисфагии. У некоторых больных поражаются дыхательные мышцы, что приводит к тяжелым дыхательным нарушениям. Дистальные мышцы конечностей поражаются сравнительно редко. Атрофия мышц и контрактуры могут развиться на поздних стадиях заболевания. Примерно у 35% больных имеются поражения сердечной мышцы, проявляющиеся инфарктоподобными изменениями на ЭКГ, аритмиями и сердечно-сосудистой недостаточностью. У некоторых больных развиваются артралгии и синдром Рейно.
Первичный дерматомиозит. Составляет треть случаев дерматомиозита-полимиозита. Поражение мышц развивается у большинства больных одновременно с изменениями кожи. На коже появляются локальные или диффузные эритематозные участки с характерным лиловым оттенком, шелушением и атрофией. Высыпания чаще всего локализуются на веках, переносице, щеках, лбу, над локтевыми, коленными и пястнофаланговыми суставами и распространяются на шею и верхнюю часть туловища. Нередко выявляется синдром Готтрона (шелушение и атрофия кожи, наиболее выраженная на кистях и стопах), гелиотропные ("солнечные") веки (припухшие и имеющие фиолетовое окрашивание) и "руки мастерового" (эритематозные гипертрофические изменения кожи ладоней и пальцев).
Поражения суставов. Иногда у больных развиваются симметричные воспалительные поражения мелких суставов кистей и лучезапястных суставов. Но деструктивные изменения суставов обычно отсутствуют. Воспаление носит преходящий характер и быстро купируется при лечении глюкокортикоидами.
Поражения легких. Чаще всего проявляются интерстициальными пневмониями, альвеолитом и медленнопрогрессирующим фиброзом, обусловленными антителами к синтетазам транспортных РНК. Проявляются эти поражения непродуктивным кашлем и прогрессирующей дыхательной недостаточностью. Поражение диафрагмальных мышц и развивающаяся сердечная недостаточность усугубляют смешанную одышку.
Поражение почек. Развивается редко и проявляется протеинурией и нефротичексим синдромом. Иногда вследствие выраженной миоглобинурии развивается острая почечная недостаточность.
Дерматомиозит и полимиозит при злокачественных новообразованиях. Примерно у 10–25% больных выявляют онкологические заболевания, главным образом, у взрослых старше 40 лет. Особенно высок риск у онкологических больных старше 60 лет. Миопатии чаще всего развиваются при раке легкого, ЖКТ, молочной железы, матки, яичников и лимфопролиферативных заболеваниях. Данные формы дерматомиозита–полимиозита рассматривают как самостоятельные и относят к паранеопластическим синдромам. Для исключения злокачественных новообразований проводят тщательное клиническое, гинекологическое, ректальное и по показаниям рентгенологическое исследование, общий и биохимический анализ крови, цитологическое исследование мокроты и мочи. Дополнительно рекомендуют определение простатспецифического антигена у мужчин и СА-125 у женщин (антиген опухоли яичника). Прогноз неблагоприятный, больные погибают от основного заболевания, осложнений или не поддающегося лечению поражения мышц.
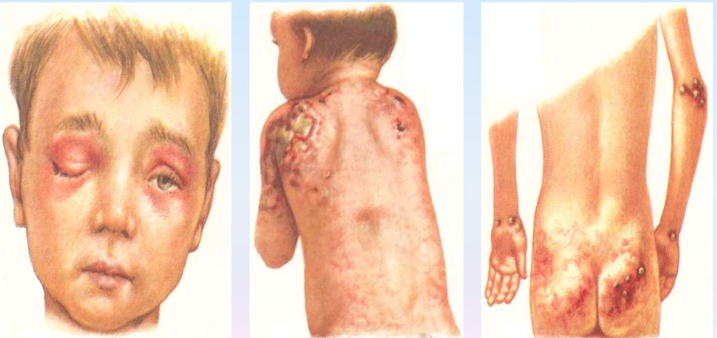
Ювенильный дерматомиозит. Среди больных дерматомиозитом примерно 25% составляют дети, у которых наблюдается клиническая и гистологическая картина васкулита с поражением кожи, мышц, ЖКТ, почек и других органов. Нередко развиваются очаги некроза кожи, ишемические инфаркты мышц, почек и ЖКТ. При прогрессировании заболевания развиваются контрактуры вследствие кальцификации мышц и околосуставной подкожной клетчатки. Прогноз заболевания такой же, как у взрослых. Но существует мнение, что у детей прогноз лучше, чем у взрослых. Возможно, эта противоречивость обусловлена различием диагностических критериев ювенильного дерматомиозита.
Миозит с "включениями". Данная форма относится к воспалительным миопатиям, носит спорадический характер и по клинике напоминает полимиозит. Болезнь развивается обычно у пожилых мужчин, проявляется очаговым порождением как проксимальных, так и дистальных мышц конечностей и имеет медленное прогрессирование. Характерно для миозита с "включениями" раннее появление выраженной слабости четырехглавой мышцы, сгибателей пальцев и запястья. Типичный гистологический признак, выявляемый в биоптате мышц, – вакуоли, окруженные ободком из базофильных гранул, содержащих амилоид. С помощью электронной микроскопии обнаруживают в саркоплазме и ядрах включения, напоминающие по форме нуклеокапсиды парамиксовирусов. В сыворотке крови отсутствуют аутоантитела. Характерна резистентность к глюкокортикоидам и другим иммуносупрессивным препаратам. Через 5–10 лет при хроническом прогрессировании течения больные утрачивают способность к самостоятельному передвижению.
Дерматомиозит и полимиозит, ассоциированный с ревматическими болезнями. Поражение проксимальных мышц, идентичное полимиозиту, может быть при системной склеродермии, ревматоидном артрите, системной красной волчанке и реже при узелковом периартериите и ревматизме. Но чаще всего миозит является компонентом смешанного заболевания соединительной ткани. На долю этой формы дерматомиозита–полимиозита приходится 20% всех перекрестных синдромов, для которых характерны полиартрит и феномен Рейно, высокий титр антинуклеарных аутоантител и повышение активности КФК. Биопсия мышц не всегда позволяет привести дифференциальную диагностику, так как выявляемые патологические изменения в мышцах сходны при полимиозите и других заболеваниях соединительной ткани. Как правило, хорошо поддается лечению глюкокортикоидами.
Диагностика
Для всех форм полимиозита характерно повышение активности мышечных ферментов (КФК, альдолазы, ЛДГ, АСЛ и АЛТ), выраженность которой коррелирует со степенью активности заболевания. Показатели общего анализа крови нормальные. У большинства (80–90%) больных выявляют аутоантитела к ядерным и цитоплазматическим антигенам. Наличие аутоантител к НEр-2 клеткам является стандартным диагностическим тестом.
Все воспалительные миопатии сопровождаются образованиями специфических аутоантител. К ним относятся аутоантитела к синтетазам транспортной РНК (антитела J0-1 к гистидил-тРНК-синтетазе) и распознающим сигналы частицам, взаимодействующим с нуклеопротеинами миозина. При сочетании полимиозита с системной склеродермией обнаруживают антитела к Sel-70, a с СКВ и синдромом Шёгрена – к Sm, Ro/SS-A La/SS-В.
При регистрации электромиограмм у большинства больных (80%) выявляют типичную миопатическую триаду: мелкие, короткие и полифазные (более чем из четырех фаз) потенциалы действия двигательных единиц. При гистологическом исследовании биоптата мышц выявляют очаговый некроз, дегенерацию и фагоцитоз мышечных волокон и воспалительные инфильтраты, состоящие из лимфоцитов, макрофагов и плазматических клеток. В скелетных мышцах развивается регенерация с базофилией мышечных элементов, увеличением числа и величины мышечных ядер. По периферии мышечных пучков развивается атрофия мышечных волокон, обусловленная некрозом эндотелия и уменьшением количества капилляров. При хроническом течении заболевания формируются интерстициальные фиброзные инфильтраты.
Диагностические критерии были разработаны A. Bohan et J.B. Petter в 1975 г. Эти критерии следует дополнить пунктами о наличии недеструктивного артрита и обнаружении аутоантител J0-1. При обнаружении одного типа поражения кожи и четырех других признаков из пунктов 1–4 и аутоантител J0-1 диагноз будет достоверен (чувствительность – 98,9%, специфичность – 95,2%).
Диагностические критерии дерматомиозита–полимиозита (по А. Bohan, J.B. Petter, 1975):
- Симметрическая слабость мышц плечевого и тазового пояса и передних сгибателей шеи, прогрессирующая в течение нескольких недель или месяцев в сочетании или при отсутствии дисфагии или поражения дыхательной мускулатуры.
- При гистологическом исследовании мышц признаки некроза мышечных фибрилл 1-го и 2-го типов, фагоцитоз, регенерация с базофилией, крупные ядра и ядрышки в сарколемме, перифасциальная атрофия, вариабельность размера миофибрилл, воспалительный экссудат.
- Повышение сывороточной концентрации мышечных ферментов (КФК, альдолаза, АСТ, АЛТ и ЛДГ).
- Электромиографические изменения: короткие, мелкие полифазные моторные единицы, фибрилляции и т.д.
- Дерматологические проявления, включающие гелиотропную окраску кожи век с периорбитальным отеком; чешуйчатый эритематозный дерматит на тыльной поверхности кистей, особенно над пястно-фаланговыми и проксимальными межфаланговыми суставами (симптом Готтрона) и поражение кожи над коленными, локтевыми суставами, лица, шеи, верхней половины грудной клетки.
Генерализованная мышечная слабость или слабость проксимальных мышц в сочетании с типичными кожными изменениями являются характерными признаками дерматомиозита–полимиозита. Но мышечная слабость, сопровождающаяся или не сопровождающаяся миалгиями, может быть при многих других заболеваниях.
Лечение
Основными препаратами при лечении тяжелого дерматомиозита–полимиозита являются глюкокортикоиды (преднизолон, метилпреднизолон). Начальная доза глюкокортикоидов колеблется от 1 до 2 мг/кг/сут, которую в течение 2–3 нед принимают 3 раза в день, затем – один раз в утренние часы. Улучшение наступает в течение 3–4 нед лечения, но у некоторых больных оно наступает через 3 мес. Отсутствие эффекта к этому времени являются основанием для увеличения дозы глюкокортикоидов.
После улучшения состояния больных, нормализации мышечной силы и КФК суточную дозу преднизолона уменьшают на 5 мг каждые 4 нед с обязательным регулярным контролем мышечной силы и активности КФК в сыворотке. Но следует учитывать то, что преднизолон в начале лечения может снижать активность КФК без уменьшения воспаления и инфильтрации мышц. Для уменьшения побочных эффектов больших доз глюкокортикоидов необходимо сочетать их с приемом Н 2 -блокаторов, витамина Д и препаратов кальция. После снижения суточной дозы до 40 мг переходят на прием 80 мг препарата через день. Если развивается рецидив заболевания дозу препарата вновь увеличивают до стойкого улучшения.
При неэффективности лечения в течение 3 мес высокими дозами глюкокортикоидов рекомендуют назначение азатиоприна в дозе 2–3,5 мг/кг/сут в 2–3 приема. Эффект развивается через 4–5 мес, показателем эффективности является снижение числа лимфоцитов до 750 мкл -1 . В последние годы используют метотрексат в начальной дозе 10 мг/нед с последующим ее постепенным повышением до 0,5 мг/кг/нед. Сочетанная терапия этих препаратов с глюкокортикоидами позволяет снизить дозу последних. Циклофосфамид и циклоспорин менее эффективны и их применяют при развитии интерстициального фиброза легких. Но все эти препараты угнетают кроветворение, вызывают нарушения ЖКТ и повышают риск инфекций.
По данным последних исследований, при неэффективности глюкокортикоидов улучшение может быть достигнуто с помощью в/в введения нормального иммуноглобулина в дозе 0,4 г/кг/сут в течение 5 сут. Применение плазмафереза показано больным с тяжелыми формами дерматомиозита–полимиозита, которым проводится лечение сочетанием глюкокортикоидов с азатиоприном или метотрексатом.
Очень важное значение имеет реабилитация больных с помощью физиотерапии, ЛФК и вспомогательных приспособлений. В острую фазу заболевания больной с помощью методиста выполняет пассивные упражнения и напряжение мышц, при улучшении состояния показаны изометрические и изотонические упражнения для восстановления мышечной силы.
Прогноз
Лечение большинства больных эффективно, но общая смертность при дерматомиозите-полимиозите в 4 раза выше, чем среди населения в целом. Пятилетняя выживаемость составляет 80%, у детей она еще выше.
Прогноз хуже у женщин и пожилых больных, при остром и тяжелом течении заболевания (высокая лихорадка, выраженная дисфагия, поражение внутренних органов), поздней диагностике и неадекватном лечении, наличии в сыворотке антител к J0-1 и злокачественных новообразованиях.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

Полимиозит и дерматомиозит - редкие системные ревматические заболевания, характеризующиеся воспалительными и дегенеративными изменениями мышц (полимиозит) либо мышц и кожи (дерматомиозит). Наиболее специфичным кожным проявлением является гелиотропная сыпь.
Поражения мышц симметричны и включают слабость, некоторую болезненность и последующую атрофию проксимальных мышц пояса верхних конечностей. Осложнения могут включать поражение внутренних органов и малигнизацию. Диагностика основана на анализе клинической картины и оценке нарушений со стороны мышц путем определения концентраций соответствующих ферментов, выполнения МРТ, электромиографии и биопсии мышечной ткани. При лечении используются глюкокортикоиды, иногда в комбинации с иммуносупрессантами или иммуноглобулинами, вводимыми внутривенно.
Женщины болеют в 2 раза чаще мужчин. Заболевание может встречаться в любом возрасте, но чаще выявляется в интервале от 40 до 60 лет; у детей - от 5 до 15 лет.

[1], [2], [3], [4], [5], [6], [7]
Что вызывает дерматомиозит и полимиозит?
Причиной заболевания, как предполагается, является аутоиммунная реакция на мышечную ткань у генетически предрасположенных лиц. Заболевание чаще встречается при наличии отягощенного семейного анамнеза и носителей некоторых HLA-антигенов (DR3, DR52, DR56). Возможными пусковыми факторами являются вирусные миозиты и злокачественные новообразования. Имеются сообщения о выявлении в мышечных клетках структур, подобных пикорнавирусам; кроме того, вирусы могут индуцировать сходные заболевания у животных. Ассоциация злокачественных опухолей с дерматомиозитом (значительно реже, нежели с полимиозитом) позволяет предположить, что опухолевый рост также может являться пусковым механизмом развития заболевания в результате запуска аутоиммунных реакций на общие антигены опухоли и мышечной ткани.
В стенках кровеносных сосудов скелетных мышц обнаруживаются отложения IgM, IgG и третьего компонента комплемента; это особенно характерно при дерматомиозите у детей. У больных полимиозитом возможно развитие также других аутоиммунных процессов.
Патофизиология дерматомиозита и полимиозита
Патологические изменения включают повреждение клеток и их атрофию на фоне воспаления различной степени выраженности. Мышцы верхних и нижних конечностей, а также лица повреждаются в меньшей степени, нежели другая скелетная мускулатура. Поражение висцеральной мускулатуры глотки и верхних отделов пищевода, реже - сердца, желудка или кишечника может приводить к нарушению функций указанных органов. Высокие концентрации миоглобина, обусловленные рабдомиолизом, могут являться причиной поражения почек. Также могут иметь место воспалительные изменения в суставах и легких, особенно у пациентов, у которых обнаруживаются антисинтетазные антитела.
Симптомы дерматомиозита и полимиозита
Начало полимиозита может быть острым (особенно у детей) или подострым (чаще у взрослых). Острая вирусная инфекция иногда предшествует или является пусковым фактором манифестации заболевания, наиболее частыми проявлениями которой являются слабость проксимальных мышц или кожные высыпания. Болевые ощущения выражены в меньшей степени, нежели слабость. Возможно развитие полиартралгии, феномена Рейно, дисфагии, нарушений со стороны легких, общих симптомов (повышения температуры тела, снижения его массы, слабости). Феномен Рейно часто встречается у больных, имеющих сопутствующие заболевания соединительной ткани.
Мышечная слабость может прогрессировать в течение нескольких недель или месяцев. Однако для клинического проявления мышечной слабости необходимо поражение минимум 50 % мышечных волокон (таким образом, наличие мышечной слабости свидетельствует о прогрессировании миозита). Пациенты могут испытывать затруднения при подъеме рук выше уровня плеч, ходьбе вверх по лестнице, подъеме из положения сидя. Вследствие выраженной слабости мышц таза и плечевого пояса пациенты могут быть прикованы к креслу-каталке или постели. При поражении сгибателей шеи становится невозможным оторвать голову от подушки. Поражение мышц глотки и верхних отделов пищевода приводит к нарушению глотания и регургитации. Мышцы нижних, верхних конечностей и лица обычно не поражаются. Однако возможно развитие контрактур конечностей.
Кожные высыпания, отмечающиеся при дерматомиозите, обычно имеют темный цвет и эритематозный характер. Характерен также периорбитальный отек пурпурного цвета (гелиотропная сыпь). Кожные высыпания могут немного возвышаться над уровнем кожи и быть гладкими или покрытыми чешуйками; локализация высыпаний - лоб, шея, плечи, грудь, спина, предплечья, нижние отделы голеней, брови, коленные области, медиальные лодыжки, тыльные поверхности межфаланговых и пястно-фаланговых суставов, с латеральной стороны (симптом Готтрона). Возможна гиперемия основания или периферии ногтей. На коже латеральной поверхности пальцев возможно развитие десквамативного дерматита, сопровождающегося появлением трещин. Первичные кожные поражения чаще разрешаются без последствий, но могут приводить к развитию вторичных изменений в виде темной пигментации, атрофии, рубцов или витилиго. Возможно образование подкожных кальцинатов, в особенности у детей.
Приблизительно у 30 % пациентов развиваются полиартралгии или полиартриты, часто сопровождающиеся отеком и суставным выпотом. Тем не менее выраженность суставных проявлений невелика. Более часто они имеют место при выявлении у больных антител к Jo-1 или другим синтетазам.
Поражение внугренних органов (за исключением глотки и верхних отделов пищевода) при полимиозите встречается реже, чем при других ревматических заболеваниях (в частности, СКВ и системной склеродермии). Редко, особенно при антисинтетазном синдроме, болезнь манифестирует в виде интерстициального пневмонита (в виде диспноэ и кашля). Могут развиваться аритмии сердца и нарушения проводимости, но они обычно бессимптомны. Проявления со стороны желудочно-кишечного тракта чаще встречаются у детей, страдающих также васкулитом, и могут включать рвоту с примесью крови, мелену и перфорацию кишечника.
Где болит?
Классификация полимиозита
Выделяют 5 вариантов полимиозитов.
- Первичный идиопатический полимиозит, который может встречаться в любом возрасте. При нем не наблюдается поражения кожи.
- Первичный идиопатический дерматомиозит подобен первичному идиопатическому полимиозиту, но при нем отмечается поражение кожи.
- Полимиозиты и дерматомиозиты, ассоциированные со злокачественными новообразованиями, могут встречаться у пациентов любого возраста; наиболее часто наблюдается их развитие у пожилых пациентов, а также у пациентов с другими заболеваниями соединительной ткани. Развитие злокачественных новообразований может наблюдаться как в течение 2 лет до, так и в течение 2 лет после дебюта миозита.
- Детский полимиозит или дерматомиозит ассоциируется с системными васкулитами.
- Полимиозит и дерматомиозит могут иметь место также у пациентов, страдающих другими заболеваниями соединительной ткани, наиболее часто - при прогрессирующем системном склерозе, смешанном заболевании соединительной ткани и СКВ.
Включение в группу полимиозитов миозита мышц туловища является некорректным, поскольку последнее представляет собой отдельное заболевание, характеризующееся клиническими проявлениями, сходными с таковыми хронического идиопатического полимиозита. Однако он развивается в пожилом возрасте, часто поражает мышцы дистальных отделов тела (например, верхних и нижних конечностей), имеет большую продолжительность, хуже отвечает на лечение и характеризуется типичной гистологической картиной.
[8], [9], [10], [11], [12], [13], [14]
Диагностика дерматомиозита и полимиозита
Полимиозит следует заподозрить при наличии у пациентов жалоб на слабость проксимальных мышц, сопровождающуюся их болезненностью или без таковой. Обследование на предмет дерматомиозита необходимо пациентам, предъявляющим жалобы на высыпания, напоминающие гелиотропные, или симптом Готтрона, а также у больных, имеющих проявления полимиозита, сочетающиеся с любыми кожными поражениями, соответствующими дерматомиозиту. Клинические проявления полимиозита и дерматомиозита могут напоминать таковые системного склероза или, менее часто, - СКВ или васкулита. Достоверность диагноза повышается при соответствии максимально большему количеству из приведенных пяти критериев:
- слабость проксимальных мышц;
- характерные кожные высыпания;
- повышение активности ферментов мышечной ткани (креатинкиназы или, при отсутствии повышения ее активности, аминотрансфераз или альдолазы);
- характерные изменения при миографии или МРТ;
- характерные гистологические изменения в биоптате мышечной ткани (абсолютный критерий).
Биопсия мышц позволяет исключить некоторые клинически сходные состояния, такие как миозит мышц туловища и рабдомиолиз, обусловленный вирусной инфекцией. Изменения, выявляемые при гистологическом исследовании, могут быть различными, однако типичными являются хроническое воспаление, очаги дегенерации и регенерации мышц. Перед началом потенциально токсичного лечения необходима установка точного диагноза (обычно путем гистологической верификации). При МРТ возможно выявление очагов отека и воспаления в мышцах с последующей прицельной их биопсией.
Лабораторные исследования позволяют подкрепить или, напротив, устранить подозрение на наличие заболевания, а также полезны при оценке его тяжести, возможности сочетания с другой аналогичной патологией и диагностике осложнений. Несмотря на то что антинуклеарные антитела выявляются у некоторых пациентов, этот феномен более характерен для других заболеваний соединительной ткани. Около 60 % больных имеют антитела к антигену ядер (РМ-1) или целых клеток тимуса и Jo-1. Роль аутоантител в патогенезе заболевания остается неясной, хотя известно, что антитела к Jo-1 являются специфичным маркером антисинтетазного синдрома, включающего фиброзирующий альвеолит, фиброз легких, артриты и феномен Рейно.
Периодическая оценка активности креатинкиназы полезна с целью мониторинга лечения. Тем не менее при выраженной атрофии мышц активность фермента может быть нормальной, несмотря на наличие хронического активного миозита. Данные МРТ, биопсии мышц или высокие значения активности креатинкиназы часто помогают дифференцировать рецидив полимиозита и миопатию, индуцированную глюкокортикоидами.
Поскольку у многих пациентов отмечаются недиагностированные злокачественные новообразования, некоторые авторы рекомендуют проведение скрининга всех взрослых, больных дерматомиозитом, и лиц, страдающих полимиозитом, в возрасте старше 60 лет по следующей схеме: физикальный осмотр, вкпючающий обследование молочных желез, гинекологическое обследование и обследование прямой кишки (в том числе исследование кала на скрытую кровь); клинический анализ крови; биохимические исследования крови; маммография; определение раковоэмбрионального антигена; общий анализ мочи; рентгенография органов грудной клетки. Необходимость проведения такого скрининга пациентам более молодого возраста, не имеющим клинических признаков злокачественных опухолей, некоторыми авторами подвергается сомнению.
[15], [16], [17], [18], [19], [20]

Общие сведения
Дерматомиозит или по-другому — болезнь Вагнера является тяжелым диффузным прогрессирующим системным заболеванием соединительной ткани, поражающим скелетную и гладкую мускулатуру, микроциркуляторную систему сосудов, внутренние органы и кожные покровы. Приводит к замене функциональных структур на фиброзные.
Клиническая картина выражается в виде двигательной дисфункции, васкулита, отеков, эритемы, гиперкератоза и может быть:
- острой, начинающейся с лихорадки и запоминающегося развития симптомов, без лечения приводит к летальному исходу спустя 2-3 мес., вызванному полным обездвиживанием и тяжелой сердечно-легочной недостаточностью;
- подострой– с постепенным нарастанием на протяжении нескольких лет мышечной слабости, артралгии, эритемы, изменений во внутренних органах;
- хронической– с доброкачественным исходом, имеющим циклическое течение и длительные ремиссии, отличается наличием локальных атрофических и склеротических изменений мышц и кожных покровов, причем во внутренних органах происходят компенсированные изменения.
В результате дерматополимиозит приводит к осложнениям – фиброзу, кальцинозу, атрофии и гнойным инфекциям.
Болезнь Вагнера встречается редко – 2-6 взрослых особей и 1-2 ребенка на 100 тыс. человек. Патология более распространена в странах на юге Европы. Заболеваемость повышается в весенне-летний сезон, что связывают с инсоляцией — более интенсивным облучением солнечным светом.
Патогенез
Начинается патогенетический процесс с неспецифического воздействия — переохлаждения, избыточной солнечной инсоляции, вакцинации, острой интоксикации, травм и прочих экзогенных факторов. Он связан с наличием инфекционных агентов, внедренных в геном мышечных клеток, изменениями нейроэндокринной реактивности или генетической предрасположенностью — наличием антигенов гистосовместимости HLA, связанных с B8 либо DR3.
При дерматомиозите происходит развитие иммунновоспалительной реакции, направленной на деструкцию измененных внутриядерных структур клеток мускулатуры и кожи. Благодаря перекрестным реакциям в иммунное поражение вовлекаются родственные по антигенам клеточные популяции. Микрофагальные механизмы элиминации из организма иммунокомплексов приводят к активации фиброгенеза и системного воспаления мелких сосудов. Гиперреактивность иммунной системы направлена на разрушение внутриядерного строения вириона, она проявляется наличием в кровотоке антител Mi2, Jo1, SRP, а также аутоантител к ядерным белкам и другим антигенам растворимого типа.
Классификация
Примерно в 25-30% случаев отсутствуют кожные симптомы и тогда говорят о частном случае этого системного заболевания – полимиозите.
В зависимости от происхождения и течения выделяют:
- первичный идиопатический дерматополимиозит;
- вторичный паранеопластический или опухолевый тип встречается у особ старше 55 лет на фоне рака молочной железы, легких, яичников, предстательной железы, матки, желудка, толстой кишки, при гемобластозах и прочих злокачественных новообразованиях;
- ювенильный – детский;
- полимиозит, сочетающийся с прочими диффузными заболеваниями соединительных тканей.
Отличается очаговым кальцинозом таких мягких тканей как мышцы, эпидермис и подкожная жировая клетчатка. Депозиты солей кальция – гидроксиапатиты могут откладываться на поверхности и глубоко, образуя ограниченные единичные или диффузно расположенные узелки и псевдоопухоли. Поверхностные бляшки с кальцинатами вызывают воспалительные реакции окружающих тканей, они нагнивают и отторгаются в виде крошковатой массы.
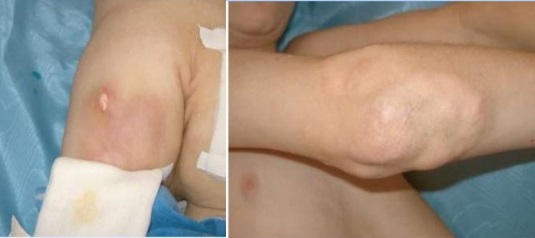
Как и у взрослых наблюдаются классические высыпания и симптом Готтрона. Постепенно нарастающая мышечная слабость начинается с причудливой походки ребенка, которая в дальнейшем еще больше сковывает движения, не давая возможности играть, бегать и обслуживать себя.
Причины
Патология полиэтиологическая, ей предшествует множество факторов:
- латентные инфекционные агенты: вирусы гриппа, парагриппа, гепатита B, пикорнавирусы, парвовирусная инфекция, бактериологические возбудителиболезни Лайма и бета-гемолитические стрептококкигруппы А, простейшие, например, токсоплазмы;
- злокачественные новообразования;
- вакцинирование от тифа, кори, паротита, холеры, краснухи;
- терапия Д-пеницилламинами, гормонами роста.
Симптомы дерматомиозита
Болезнь Вагнера представляет собой целый симпатокомплекс, обусловленный прежде всего генерализованными нарушениями микроциркуляторного кровяного русла и проявляющийся со стороны различных органов и систем, больше всего – со стороны эпидермиса и мускулатуры.
Симптом Готтрона – наличие розовых и ярко-красных шелушащихся папул, бляшек, локализованных на разгибательных поверхностях суставов, таких как: межфаланговые и пястнофаланговые складки, локтевые и коленные сгибы. Неяркие покраснения могут самопроизвольно регрессировать и оказываться полностью обратимыми.
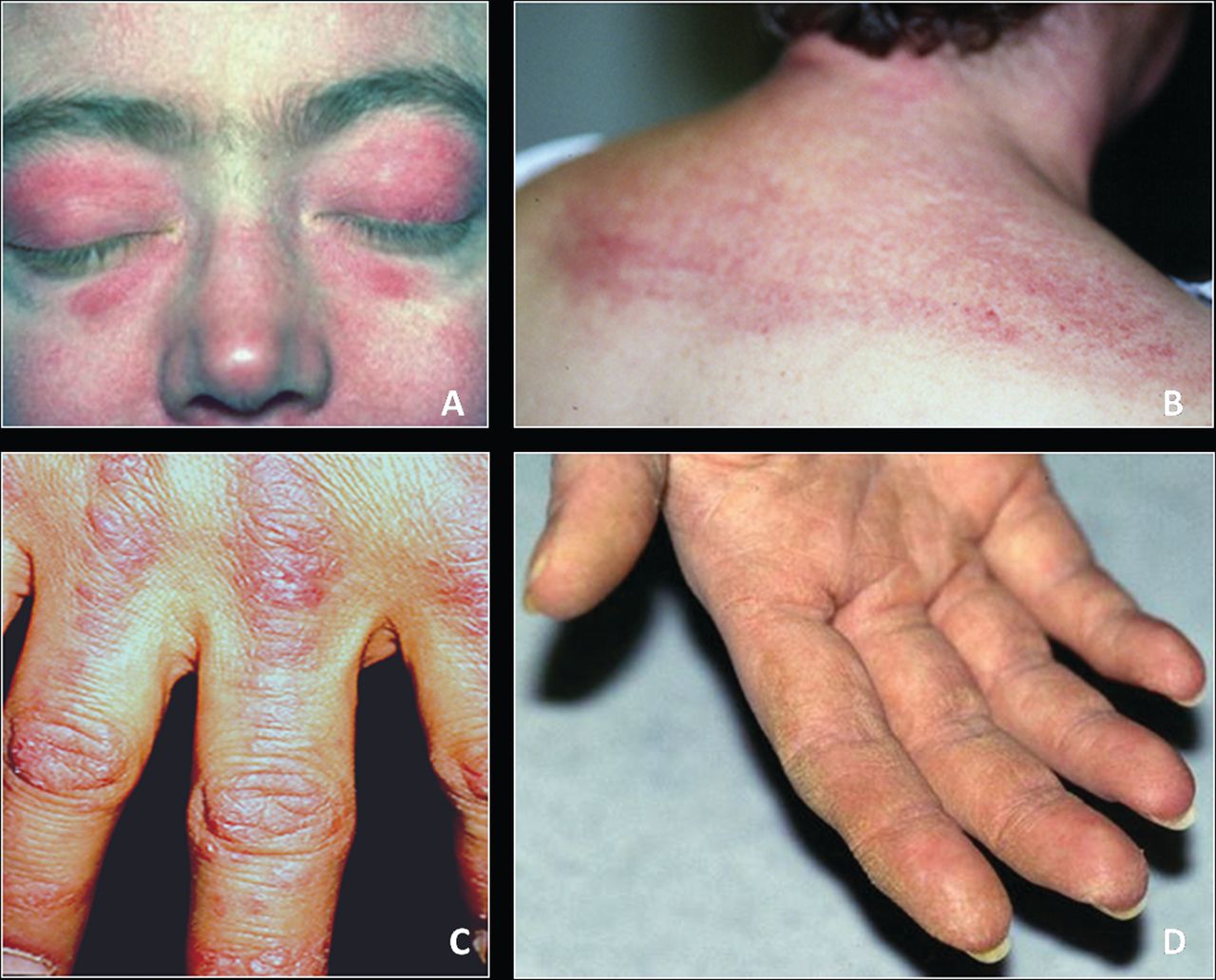
У больных развивается фотодерматит – повышение чувствительности к солнечным лучам открытой поверхности кожи.
Ранним симптомом дерматомиозита считается изменения ногтей – покраснение и разрастание кожи вокруг ногтевой пластинки.
Главные симптомы полимиозита – это атрофия и мышечная слабость, которая обычно симметрична и вовлекает в патологический процесс плечевой и тазовый пояс, сгибатели шеи и мышцы брюшного пресса, проявляется сложностью подъема по лестнице, вставания со стула, удержания головы.
Опаснее всего, когда задеты дыхательные, глазодвигательные и глотательные мышцы, ведь это провоцирует дыхательную недостаточность, дезориентацию и диплопию, трудности во время акта глотания (дисфагия), охриплость и изменения голоса. Воспалительный процесс в мускулатуре может сопровождаться болевым синдромом — миалгией. Нарушения кровоснабжения и трофики вызывает уменьшение мышечной массы, развитие тяжелого некротического миозита, в результате происходит разрастание соединительной ткани в мышечных волокнах и образование уплотнений, а также сухожильно-мышечных контрактур.
Влияние на сердечно-сосудистую систему вызывает не только патологию миокарда — миокардит, но и коронарных сосудов и всех трех оболочек сердца, провоцируя тахикардию, артериальную гипотензию, приглушенность тонов, дилатационную кардиопатию, нарушения сердечного ритма, вплоть до AV-блокады и инфаркта.
Обычно это вторичные осложнения, вызванные гиповентиляцией в результате слабости дыхательных мышц и диафрагмы, развитием инфекции, аспирацией при глотании. Чаще всего они приводят к фиброзирующему альвеолиту, базальному пневмофиброзу, интерстициальной пневмонии и даже метастатическим новообразованиям. Главными проявлениями являются такие симптомы как:
- одышка;
- хрипы;
- непродуктивный кашель;
- цианоз;
- крепитация– хрустящий звук, выявляющийся при аускультации;
- уменьшение объемов и диффузионных характеристик легких.
- скованность движений – вследствие поражения суставов ( артралгии и артрита), что может сопровождаться их припухлостью и болями;
- субфебрильная температура – до 38° в острых фазах;
- развитие гастрита, колита, эрозивно-язвенных поражений и геморрагиипрактически в 50% случаев приводят к утрате веса и спровоцированы изменениями сосудов и нарушениями питания слизистых ЖКТ, проходимости нервных импульсов и перистальтики;
- синдром Рейно – онемение рук и холодные пальцы после действия холода, эмоционального стрессалибо без явных причин, что в итоге вызывает боль и отечность, багрово-красный цвет кожных покровов;
- конъюнктивит, стоматит и другие поражения слизистых;
- умеренная гепатомегалия и незначительные нарушения функции печени;
- лимфаденопатия– наличие плотных распространенных отеков;
- нарушения чувствительности — гиперестезии, гипералгезии, арефлексии, чаще всего вызванные полиневритом;
- гормональный дисбаланс, сопровождающийся функциональными нарушениями работы половых желез и гипофизарно-надпочечниковой системы.
Анализы и диагностика
При подозрении на дерматомиозит необходим первичный осмотр и изучение состояния больного, сбор данных анамнеза и жалоб. В анализах крови можно выявить:
- незначительное повышение СОЭ;
- умеренный лейкоцитоз;
- увеличенное количество миоглобина, фибриногена, креатина, мочевой кислоты, гаптоглобинов, серомукоида, α-2-иγ-глобулинов;
- повышение ферментативной активности, вызванной мышечным распадом, наибольшее диагностическое значение имеет креатинфосфокиназа, лактатдегидрогеназа, АЛТ, АСТ, альдолаза.
При полимиозите важны электромиографические исследования нормальной электрической активности расслабленных мышц, низкоамплитудной — при их произвольных сокращениях, а также изучение коротких, полифазных потенциалов моторных единиц, выявляющих спонтанные потенциалы фибрилляции.
Для подтверждения диагноза следует провести:
- биопсию мышц;
- иммунологические пробы антител Jo 1 к гистидил-транспортной РНКсинтетазе;
- рентгенографию и УЗИ для выявления альвеолита, пневмонии, кальцинатов, остеопорозаили других глубинных изменений.
Лечение дерматомиозита
В основном лечение дерматомиозита медикаментозное. Обычно для ликвидации иммунновоспалительного фиброзирующего процесса назначают:
- глюкокортикоиды;
- цитостатики.
Кроме того, лечение полимиозита может включать симптоматическую коррекцию нарушений работы органов и систем – прием сердечных гликозидов, гепатопротекторов, поливитаминных комплексов, нестероидных противовоспалительных препаратов и средств, улучшающих нервно-мышечную передачу и повышающих метаболические процессы в мышечных тканях. При обнаружении глубинного или поверхностного отложения кальцинатов вводят внутривенно с 5%-раствором глюкозы динатриевую соль этилендиаминтетрауксусной кислоты, которая образует комплексы с ионами кальция, и они выводятся из организма.
В ходе лечения важно сохранять объем движений и препятствовать мышечной слабости, образованию контрактур, к примеру, при помощи лечебной физкультуры. Больным категорически противопоказана иммобилизация.
Читайте также: