
Генетическая болезнь атрофирование мышц
Существует огромное количество различных заболеваний, которые возникают у деток независимо от обстоятельств или действия окружающей среды. Это категория именно наследственных болезней. Сейчас же пойдет речь о такой проблеме, как мышечная дистрофия Дюшенна: что это за хворь такая, каковы у нее симптомы и можно ли с ней справиться.
Терминология
Изначально нужно узнать, что же такое наследственные болезни. Так, это заболевания, которые возникают в результате дефектов аппарата наследственных клеток. То есть это определенные сбои, которые происходят на генетическом уровне.
Мышечная дистрофия Дюшенна – это именно наследственная болезнь. Проявляется она очень быстро, основной симптом в данном случае – это быстро прогрессирующая слабость в мышцах. Нужно отметить: как и все остальные мышечные заболевания, болезнь Дюшенна также приводит в конечном результате к атрофии мышц, нарушению моторики и, конечно же, инвалидности. В подростковом возрасте детки с таким диагнозом уже не имеют возможности самостоятельно передвигаться и не могут обходиться без посторонней помощи.
Что происходит на генном уровне
Как уже было отмечено, мышечная дистрофия Дюшенна – это генетическое заболевание. Так, происходит мутация в том гене, который отвечает за выработку особого белка дистрофина. Именно он и необходим для нормальной работы мышечных волокон. При этом важно отметить, что эта генетическая мутация может как передаваться по наследству, так и возникать спонтанно.
Также важно отметить, что ген локализируется в хромосоме Х. Но женщины этой болезнью заболеть не могут, являясь только лишь передатчиком мутации от поколения к поколению. То есть если мама передаст мутацию сыну, он с 50%-й вероятностью заболеет. Если же девочке, она просто будет носителем гена, клинических проявлений болезни у нее не будет.
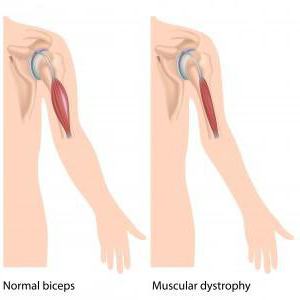
Симптоматика: группы
В основном, болезнь активно заявляет о себе примерно в 5-6 летнем возрасте. Однако первые симптомы могут возникнуть у малыша, который еще не достиг трехлетнего возраста. При этом надо отметить, что все патологические нарушения медки условно разделяют на несколько больших групп:
- Поражение мускулатуры.
- Поражение сердечной мышцы.
- Деформация скелета ребенка.
- Различные эндокринные расстройства.
- Нарушения нормальной умственной деятельности.
Наиболее часто встречающиеся проявления болезни
Обязательно также надо рассказать о том, как проявляется синдром Дюшенна. Симптомы бывают следующие:
- Слабость. Которая постепенно нарастает, развивается.
- Начинается прогрессирующая мышечная слабость именно с верхних конечностей, далее затрагиваются ноги и только потом – все остальные части тела и органы.
- Ребенок утрачивает возможность сам передвигаться. Примерно к 12-летнему возрасту такие детки уже полностью зависимы от инвалидной коляски.
- Также наблюдаются расстройства дыхательной системы.
- Ну и, конечно же, бывают нарушения в работе кардиологической системы. Позже происходят необратимые изменения в миокарде.
О поражении мышц скелета
Именно поражения мышечной ткани – наиболее распространенный симптом, если речь идет о такой проблеме, как синдром Дюшенна. При этом надо отметить, что рождаются детки без особых отклонений в развитии. В малом возрасте ребята менее активны и подвижны, нежели сверстники. Но чаще всего это связывают с темпераментом и характером ребенка. Поэтому отклонения очень редко замечаются. Более существенные признаки проявляются уже во время ходьбы малыша. Такие детки могут передвигаться на носочках, не становясь на полную стопу. Также они частенько падают.
Особым показателем является еще и симптом Говерса. То есть ребенок, чтобы подняться с пола, активно пользуется руками, как бы взбираясь по самому себе.
Также надо отметить, что при такой проблеме, как синдром Дюшенна, у ребенка постепенно атрофируются мышцы. Но нередко бывает так, что у крохи внешне мускулатура кажется очень развитой. Мальчик даже на первую вскидку оказывается как бы накачанным. Но это только лишь обман зрения. Все дело в том, что в процессе болезни мышечные волокна постепенно распадаются, а их место занимает жировая ткань. Отсюда и такой внушительный внешний вид.

Немного о деформации скелета
Если у ребенка прогрессирующая мышечная дистрофия Дюшенна, то постепенно у мальчика изменится форма скелета. Сначала патология затронет поясничный отдел, далее возникнет сколиоз, то есть произойдет искривление грудного отдела позвоночника. Позже проявится сутулость и, конечно же, изменится нормальная форма стопы. Вся данная симптоматика еще в большей степени будет сопутствовать ухудшению двигательной активности малыша.
О сердечной мышце
Обязательным симптомом при данной болезни также является поражение сердечной мышцы. Происходит нарушение ритма сердца, возникают регулярные перепады артериального давления. При этом сердце увеличивается в размерах. Но его функциональные возможности наоборот, уменьшаются. И в результате постепенно формируется сердечная недостаточность. Если эта проблема еще будет сочетаться с дыхательной недостаточностью, то возникает большая вероятность летального исхода.

Нарушения умственной активности
Нужно отметить, что мышечная дистрофия Дюшенна-Беккера не всегда проявляется таким симптомом, как умственная отсталость. Связано это может быть с дефицитом такого вещества, как аподистрофин, необходимого для работы головного мозга. Нарушения интеллекта могут быть самыми разными – от слабой умственной отсталости до идиотии. Усугублению этих когнитивных расстройств способствует еще и невозможность посещать садики, школы, кружки и иные места скопления детей. В результате возникает социальная дезадаптация.
Расстройства работы эндокринной системы
Различные эндокринные расстройства встречаются не более чем у 30-50% всех больных. Чаще всего это именно лишний вес, ожирение. При этом детки также имеют более низкий, чем у сверстников, рост.
Исход болезни
Какова клинико-эпидемиологическая характеристика мышечной дистрофии Дюшенна? Так, частота возникновения болезни – 3,3 пациента на 100 тысяч здоровых людей. Нужно отметить, что мышечная атрофия постепенно прогрессирует, и к 15-летнему возрасту мальчик уже не может обходиться без помощи окружающих, являясь полностью обездвиженным. Ко всему, происходит еще и частое присоединение различных бактериальных инфекций (чаще всего именно мочеполовой и дыхательной систем), при неправильном уходе за ребенком возникают пролежни. Если проблемы с дыхательной системой соединяются с сердечной недостаточностью, это грозит смертельным исходом. Если же говорить в общем, то такие пациенты практически никогда не живут более 30 лет.
Диагностика болезни
- Генетическое тестирование, то есть анализ ДНК.
- Электромиография, когда подтверждается первичное изменение мышц.
- Биопсия мышц, когда происходит определение наличия белка дистрофина в мышце.
- Анализ крови на определение уровня креатинкиназы. Нужно отметить, что именно этот фермент указывает на гибель мышечных волокон.
Лечение
Полностью излечиться от данной болезни невозможно. Можно только облегчить проявление симптомов, что сделает жизнь больного немного проще и удобнее. Так, после того, как пациенту ставят такой диагноз, чаще всего ему назначают терапию глюкокортикостеродами, которые призваны замедлить процесс развития болезни. Иные процедуры, которые также могут быть использованы при данной проблеме:
- Дополнительная вентиляция легких.
- Терапия медикаментами, которая направлена на нормализацию работы сердечной мышцы.
- Использование различных приспособлений, которые повышают мобильность пациента.
Также важно отметить, что сегодня ведутся разработки новейших методик, которые основаны на генной терапии, а также пересадке стволовых клеток.

Иные мышечные заболевания
Существуют еще и иные мышечные врожденные заболевания детей. К таким болезням можно отнести, помимо дистрофии Дюшенна:
- Дистрофию Беккера. Эта болезнь очень похожа на синдром Дюшенна.
- Мышечную дистрофию Дрейфуса. Это медленно прогрессирующая болезнь, при которой интеллект сохраняется.
- Прогрессирующую мышечную дистрофию Эрба-Рота. Проявляется в подростковом возрасте, прогрессирование быстрое, инвалидизация наступает рано.
- Плечелопаточно-лицевую форму Ландузи-Дежерина, когда мышечная слабость локализируется в области лица, плеч.
При этом надо отметить, что ни при одной из этих болезней не проявляется мышечная слабость у новорожденных. Вся симптоматика возникает в основном в подростковом возрасте. Длительность жизни пациентов чаще всего не превышает 30 лет.
Спинальная мышечная атрофия относится к генетически обусловленным заболеваниям и характеризуется прогрессирующей атрофией мышц в результате нарушения периферической иннервации. При этом происходит блокада импульсов спинного мозга, идущих к мышечным волокнам, что вызывает прекращение их нормального развития и функционирования. Чаще поражаются мышцы туловища и проксимальных отделов конечностей. Болезнь развивается у новорожденных детей, но иногда поражает людей молодого возраста. 
Причины развития болезни
Развитие патологии связывают с мутацией генов, которая приводит к нарушению синтеза специфического белка, участвующего в жизнедеятельности двигательных нейронов спинного мозга. Они представляют собой скопление клеток, которые локализуются в передних спинномозговых отделах и образуют двигательные корешки. Главной функцией нейронов считается регулирование сократительной активности, кровообращения, обменных процессов мышечных волокон.
При нарушении синтеза белка мотонейроны постепенно разрушаются как в правой, так и левой половине спинного мозга. Симметричная спинная мышечная атрофия относится к важному диагностическому признаку.
В результате к мышцам не поступают сигналы из центральной нервной системы, они теряют способность к сокращению. Это негативно влияет на двигательную активность и приобретение ребенком навыков самообслуживания. При этом чувствительная иннервация сохраняется в полном объеме.
Заболевание передается по аутосомно-рецессивному типу, поэтому встречается в популяции населения крайне редко. Для того чтобы патология начала развиваться в организме, необходимо носительство дефектного гена обоими родителями. При этом чаще всего у них нет проявлений заболевания, а обоюдное носительство не всегда дает появление недуга у совместного ребенка. Доказано, что каждый 40 человек может содержать в своем генетическом материале мутации определенного вида хромосом, обуславливающие носительство дефектного гена.
Проявления заболевания
Выделяют несколько форм развития патологии, которые отличаются возрастом возникновения первых симптомов, тяжестью течения, продолжительностью жизни.
Чем позже развивается патология, тем лучше прогноз для жизни и поддержания навыков самообслуживания.
В любом случае больные становятся инвалидами, требующими дополнительный уход и специальных средств передвижения (коляски, ходунки). Часто пациенты прикованы к кровати, их состояние осложняется застойными процессами в легких, которые могут привести к летальному исходу.

Спинальная мышечная атрофия тип 1 может быть заподозрена еще во внутриутробном периоде при слабом шевелении ребенка во второй половине беременности. На протяжении полугода после рождения отмечают вялость, низкую активность новорожденного. При обследовании не обнаруживают сухожильные рефлексы (ахиллов, коленный).
Мышцы верхних и нижних конечностей, туловища и головы не развиваются. Слабость и атрофия мышечных волокон приводит к неспособности детей полноценно сидеть, ползать, вставать на ноги. По мере роста скелета возникает деформация костей. Например, при попытке новорожденного сидеть позвоночник не поддерживается мышечным каркасом спины, что приводит к развитию кифоза.
Нередко выявляют непроизвольное подергивание мышц, дрожание пальцев вытянутых рук. Мышечная атрофия может визуально сглаживаться чрезмерным развитием подкожно-жировой клетчатки. Из-за недоразвития межреберных мышц грудная клетка приобретает уплощенную форму. Страдает функция глотания, сосания, дыхания, в результате чего развиваются тяжелые осложнения: аспирационная и застойная пневмония, истощение, дыхательная недостаточность.
Развитие умственной сферы проходит удовлетворительно, так как отделы головного мозга не подвергаются патологическим изменениям.
Эта форма заболевания считается неблагоприятной для жизни, такие дети обычно погибают в течение первых лет после рождения.
Атрофия спинного мозга в случае 2 типа болезни проявляется в конце первого года жизни, обычно в 7−18 месяцев ребенка. Признаки патологии выражены не резко и развиваются постепенно. По мере взросления новорожденных отмечают слабую двигательную активность. Дети предпочитают лежать, неохотно перемещаются в пространстве, значительно отстают в физическом развитии от своих сверстников.
Мышцы тела и конечностей подвержены непроизвольным сокращениям, отмечают подергивание языка. Дети способны сидеть, вставать на ноги, иногда медленно передвигаться с посторонней помощью. Прием пищи и дыхание обычно не затруднено, сухожильные рефлексы развиты слабо. Продолжительность жизни таких больных снижают частые респираторные инфекции и тяжелые застойные воспаления легких в результате вялой двигательной активности.
Первые симптомы недуга появляются в конце второго года жизни ребенка, иногда развитие мышечной атрофии начинается в период полового созревания. Заболевание характеризуется медленным прогрессированием. Сначала атрофируются верхние мышечные группы ног, затем патологический процесс поражает руки, туловище, шею. 
Больные, страдающие 3 типом болезни, способны передвигаться: вставать, ходить, подниматься по лестнице. Характерным признаком заболевания считается гипертрофия ягодичных и икроножных мышц. Однако по мере нарастания дистрофических изменений в передних отделах спинного мозга сухожильные рефлексы угасают, что свидетельствует о необратимых изменениях мышечной ткани.
Со временем двигательная активность снижается, что заставляет больных пользоваться инвалидными колясками. Длительно сохраняется способность к самообслуживанию, тазовые функции развиты хорошо (мочеиспускание, дефекация). Чувствительная иннервация при заболевании не страдает.
Терапевтическая тактика
Лечение спинальной мышечной атрофии имеет поддерживающий характер и проводится в течение всей жизни больного.
Выздоровления достичь невозможно, а лишь замедлить развитие патологии в нервной и мышечной ткани.
Терапевтическая тактика включает комплекс мер, направленных на сохранении двигательной активности.
- Лекарственные средства, нормализующие проведение нервного импульса — прозерин, нивалин.
- Ноотропные препараты, улучшающие кровообращение нервной ткани — пирацетам, ноотропил.
- Средства, стимулирующие кровоток и обменные реакции в мышечной ткани — актовегин, оротат калия.
- Витаминные препараты, обладающие антиоксидантным действием — токоферол, витамины группы В, глутаминовая кислота.
- Физиопроцедуры, усиливающие кровообращения пораженных конечностей — парафин, УВЧ, электрофорез с прозерином.
- Массаж — стимулирует мышечную активность, способствует выведению продуктов обмена.
- Лечебная гимнастика — атрофия мышц спины, рук, ног после занятий замедляется.
- Ортопедическая помощь — использование специальных приспособлений, поддерживающих двигательную активность грудной клетки, верхних и нижних конечностей.
В терминальных стадиях заболевания появляются дыхательные расстройства, которые требуют перевода пациентов на искусственную вентиляцию легких.
Спинальная мышечная атрофия считается тяжелым генетическим заболеванием с постоянным прогрессированием патологического процесса и неблагоприятным прогнозом.
В последние годы ученые всего мира ведут научные разработки по синтезированию лекарственного препарата, способного замещать недостающий нейронный белок. Эта единственная надежда больных на выздоровление и значительное улучшение качества жизни.
Спинальная мышечная атрофия – одно из самых опасных генетически обусловленных заболеваний, которое обнаруживается у младенцев, подростков, взрослых.

Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Спинальная мышечная атрофия — что это
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
Спинальная мышечная атрофия — это разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Симптомы
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.

Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.

Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:

Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.
При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить. Рекомендовано регулярно делать массаж, физиопроцедуры. Детей даже с ограниченными движениями возят в бассейн.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:
Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015
Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
Спинальная мышечная атрофия является основной генетической причиной смерти в детском возрасте. Давайте разберемся в причинах и способах изменения жизни ребенка, а также узнаем, какие методы лечения доступны на сегодняшний день.
Что такое спинальная мышечная атрофия
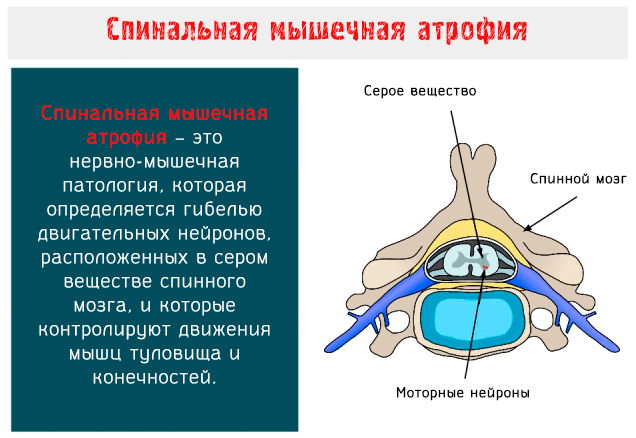
Спинальная мышечная атрофия (SMA: Spinal Muscular Atrophy) – это нервно-мышечное заболевание аутосомно-рецессивного типа, характеризуется гибелью двигательных нейронов, расположенных в переднем роге серого вещества спинного мозга и в нижней части ствола головного мозга.
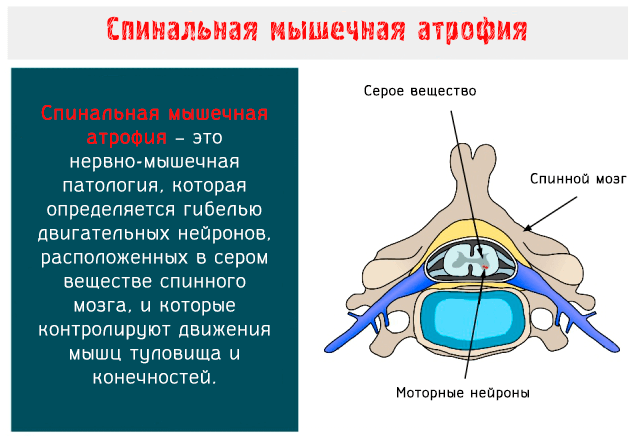
Моторные нейроны – это клетки, из которых образуются нервы, предназначенные для управления скелетными и поперечно-полосатыми мышцами глотки и гортани: когда они вырождаются, целые группы волокон подвергаются атрофии и, соответственно, результатом является мышечная слабость.
Работы глазных мышц, хотя и управляется моторными нейронами энцефального ствола мозга, не нарушается при этой болезни.
Частота спинальной мышечной атрофии колеблется от 1:6000 до 1:10000, и подвержены ей все этнические группы; является редким заболеванием, является одним из самых распространенных нервно-мышечных заболеваний, точнее вторым после дистрофии Дюшенна.
Причина спинально мышечной атрофии
Причина спинально мышечной атрофии была обнаружена в середине 90-х годов, спустя сто лет после первого описания болезни. В 95% случаев речь идёт о делеции в гене SMN1, локализованном на длинном плече хромосомы 5 (делеция – это потеря последовательности ДНК).
Поскольку спинально мышечная атрофия наследуется по аутосомно-рецессивному типу, для развития болезни человек должен получить обе копии плохого SMN1 – от матери и от отца. Таких родителей называют гетерозиготными или носителями, и они не имеют симптомов заболевания. Носители встречаются с частотой 1:50.
Ген SMN1 кодирует белок SMN, который используется в цитоплазме и ядре всех клеток и имеет решающее значение для формирования snRNP, малых ядерных рибонуклеопротеидов, компонентов сплайсинг машин.
Почему же вездесущий белок SMN является критическим фактором для выживания и надлежащего функционирования моторных нейронов?
В 2012 году Лотти и соавторы показали, что белки SMN имеют имеют важное значение для дифференциации и бесперебойной работы моторных нейронов.
Другие гипотезы были сформулированы для объяснения антиапоптической роли SMN:
- потребность в этом белке выше у моторных нейронов, чем в других тканях.
- по мнению других авторов, это можно объяснить тем, что белок SMN участвует в транспортировке вдоль аксонов РНК-связывающих белков.
Несмотря на все предположения, в настоящее время ещё неясно, какая из многих функций протеина SMN связана с развитием спинально мышечной атрофии.
4 типа спинально мышечной атрофии
Спинально мышечную атрофию классифицируют на четыре типа, в соответствии:
- с возрастом появления симптомов
- с максимальной двигательной активностью, на которую способен больной
У 25% лиц избегают точной классификации. Кроме того, у людей, страдающих от одного типа заболевания, симптомы могут существенно различаться.
Это самая тяжелая форма спинально мышечной атрофии, составляет 50% от всех случаев.
Главные её особенности:
- проявляется до 6-го месяца жизни
- ребенок имеет плохую и дряблую мышечную массу: он мало движется, потому что не может противостоять силе тяжести, не в состоянии держать голову в вертикальном положении и сидеть без поддержки
- кости хрупкие и подвержены переломам, кроме того, в позвоночнике развивается сколиоз. Проблемы с костями у пациента со спинально мышечной атрофией не удивляют, так как именно физическая активность способствует минерализации костей
- рефлекс сосания и глотания слабый, поэтому такого ребёнка трудно кормить
- грудная клетка ребенка меньше нормы из-за слабости дыхательных мышц. Кашлевый рефлекс слабый, что нарушает процесс избавления от выделений (слизи и твердые частицы, включая микробов)
У детей, страдающих от спинально мышечной атрофии 1 типа, часто развивается пневмония, так как они не в состоянии избавиться от каких-либо патогенных микроорганизмов с кашлем, а также из-за потери контроля глотательных мышц, которые не могут предотвратить попадание слюны и кусочков пищи в легкие. Повторяющие пневмонии ведут, к сожалению, к дыхательной недостаточности.
Для тех, кто страдает от этой формой патологии прогноз неблагоприятный: смерть наступает в течение 2 лет, даже самое хорошее лечение продлевает жизнь только до 5 лет.
Промежуточная форма спинальной мышечной атрофии.
Давайте посмотрим характеристики:
- проявляется между 6 и 18 месяцами
- ребенок показывает задержку в развитии моторики: не в состоянии сидеть, ему нужна поддержка, чтобы стоять, и никогда не научится ходить. Может иметь легкий тремор рук
- при этом типе также отмечается склонность к развитию сколиоза и хрупкости костей
- у некоторых маленьких пациентов дисфагия становится препятствием для поглощения достаточного для развития количества калорий
- кашлевый рефлекс может ослабнуть, облегчая возникновение респираторных инфекций
При спинальной мышечной атрофии 2 типа также высок риск развития дыхательной недостаточности. Прогрессирование симптомов настолько разнообразно, что некоторые пациенты умирают в младенчестве, другие в состоянии достичь зрелости.
Детская форма спинальной мышечной атрофии, которая:
- может возникнуть в возрасте от полутора лет
- по сравнению с предыдущими случаями, дети могут стоять и ходить самостоятельно, эта способность в некоторых случаях сохраняется до зрелого возраста
- наблюдается тремор рук и могут возникнуть проблемы с суставами и сколиоз
- нарушения дыхания и глотания проявляются менее часто, чем при 1 и 2 типе
У людей, страдающих от 3 типа спинальной мышечной атрофии, средняя продолжительность жизни сравнима со здоровыми людьми. Но, из-за проблем с питанием и низкой физической активность, часто имеют избыточный вес.
Продолжительность жизни нормальная.
Как распознать спинальную мышечную атрофию
Специалист по детской неврологии задаст ряд вопросов, чтобы получить подробный отчет о медицинской истории ребенка и его семьи, после процедуры физического обследования, чтобы оценить физическое состояние маленького пациента.
Подтверждение диагноза спинальной мышечной атрофии достигается благодаря генетическому тесту: берут образец крови и исследуют на наличие аномального гена SMN1. Тест можно использовать, чтобы найти носителей.
Поиск неисправного SMN1 также может осуществляться путём биопсии ворсинок хориона, которые являются частью плаценты, что делает возможным пренатальную диагностику в случае:
- если у пары уже был ребёнок, пострадавший от спинальной мышечной атрофии
- партнеры обнаруживают, что являются носителями, но все равно хотя родить ребёнка
Иногда бывает, что нельзя точно утверждать, что это спинальная мышечная атрофия. Тогда используют другие тесты, которые помогают провести дифференциальный диагноз между спинальной атрофией и другими патологиями нервов и мышц:
- электромиография, которая измеряет электрическую активность мышц
- мышечная биопсия, то есть изучение образцов мышечной ткани
- оценка концентрации креатина киназы, фермента уровень которого повышается при повреждении мышц
Как облегчить симптомы спинальной атрофии
На данный момент не существует лекарств для лечения спинальной мышечной атрофии, поэтому пациенты могут воспользоваться только поддерживающим лечением.
- физиотерапия
- диетология
- дыхание
Для пациентов школьного возраста важно, чтобы они активно участвовали в школьных мероприятиях, потому что их физическая инвалидность никак не влияет на способности к обучению.
Физиотерапия необходима независимо от возраста человека. Упражнения позволят максимизировать амплитуду движений, чтобы предотвратить или замедлить потерю мелкой моторики. Дети со спинальной мышечной атрофией 1 и 2 типа получают огромную пользу от гимнастики в бассейне, поскольку вода помогает стимулировать всю мышечную массу.
Пациентам с 3 типом спинальной мышечной атрофии нужны ортопедические устройства (инвалидные коляски, параподы и т.д.), которые обеспечивают удобство и мобильность. Упражнения также важны, поскольку помогают предотвратить сколиоз, который усугубляет проблемы с дыханием и движениями.
Каждый человек, страдающий от спинальной мышечной атрофии, должен иметь свой индивидуальный план питания, чтобы предотвратить последствия недостаточного или избыточного питания.
У тех детей, которые имеют большие трудности при грудном кормлении, пережевывании пищи и глотании, нужно принять меры, чтобы избежать таких осложнений, как аспирационная пневмония.
- Вы можете прибегнуть к использованию назогастрального зонда, который проходит через нос и доставляет пищу в желудок. Его относительно легко установить и снять, но он может протекать, тогда его следует заменить
- Другой вариант – гастростомия, то есть вывод трубки из желудка; является более простым в обслуживании, но процедура выполняется в операционной под наркозом.
Дыхание
Есть в этом случае для со спинальной мышечной атрофией существует три цели:
- пациенты и всех люди, которые вступают в контакт с ними, должны быть вакцинированы, например, против вируса гриппа, пневмококковой инфекции и бактерии коклюша, потому что инфекции дыхательных путей могут быть очень опасными для таких пациентов
- если кашлевый рефлекс слабым, это можно исправить с помощью специального устройства (Cough Assist): оно создает быстрое изменение давления снаружи и внутри легких, и быстрое прохождение воздуха по дыхательным путям, что имитирует кашель, освобождая дыхательные пути от секрета и микробов
- наконец, важна оценка дыхательной функции этих субъектов по степени сатурации кислорода в крови. Если количества кислорода меньше потребностей, стоит серьёзно рассмотреть идею использования механического респиратора. Изначально он используется в случае инфекции дыхательных путей и во время сна; с развитием атрфоии – весь день.
Открытие причины заболевания открыло для исследовательских групп большое направление для поиска методов лечения, направленных на максимально возможное замедление прогрессирования симптомов: повышение уровня белка SMN.
- Поскольку спинальная мышечная атрофия является моногенным заболеванием, это позволяет вмешаться в корень недуга, предоставляя пациентам функционирующий ген SMN1 (генная терапия)
- У лиц, страдающих от спинальной атрофии, но имеющих ген SMN2, можно увеличить экспрессию этого гена и заблокировать исключение экзона 7 во время сплайсинга незрелой мРНК.
В обоих случаях количество функционирующего белка SMN увеличивается.
AVXS-101 – экспериментальный препарат, разработанный биотехнологической компании Авексис, которому удалось достичь 1 этапа экспериментов на людях, при оценке безопасности лечения, и он начинает проверку эффективности.
Авексис сосредоточились на детях, страдающих от спинальной мышечной атрофии 1 типа, потому что это самый распространенный и смертельный тип заболевания.
AVXS-101 состоит из большого числа частиц адено-ассоциированного вируса серотипа 9, неспособного к репликации, но содержащего одну копию нормального гена SMN1.
Вводится в организм внутривенно, в состоянии преодолеть гематоэнцефалический барьер и достичь моторных нейронов.
Молекула ДНК, переносимая каждым вирусным вектором, производится в лаборатории. Она не изменяет ДНК пациента; содержит промотор, т.е. последовательность, которая способствует транскрипции ДНК в РНК, и гарантирует постоянное производство протеина SMN.
Анализ промежуточных данных, опубликованных Авексис в апреле 2016 года показывает, что:
Читайте также: