
Генетическая болезнь мышц тела
Спинальная мышечная атрофия относится к генетически обусловленным заболеваниям и характеризуется прогрессирующей атрофией мышц в результате нарушения периферической иннервации. При этом происходит блокада импульсов спинного мозга, идущих к мышечным волокнам, что вызывает прекращение их нормального развития и функционирования. Чаще поражаются мышцы туловища и проксимальных отделов конечностей. Болезнь развивается у новорожденных детей, но иногда поражает людей молодого возраста. 
Причины развития болезни
Развитие патологии связывают с мутацией генов, которая приводит к нарушению синтеза специфического белка, участвующего в жизнедеятельности двигательных нейронов спинного мозга. Они представляют собой скопление клеток, которые локализуются в передних спинномозговых отделах и образуют двигательные корешки. Главной функцией нейронов считается регулирование сократительной активности, кровообращения, обменных процессов мышечных волокон.
При нарушении синтеза белка мотонейроны постепенно разрушаются как в правой, так и левой половине спинного мозга. Симметричная спинная мышечная атрофия относится к важному диагностическому признаку.
В результате к мышцам не поступают сигналы из центральной нервной системы, они теряют способность к сокращению. Это негативно влияет на двигательную активность и приобретение ребенком навыков самообслуживания. При этом чувствительная иннервация сохраняется в полном объеме.
Заболевание передается по аутосомно-рецессивному типу, поэтому встречается в популяции населения крайне редко. Для того чтобы патология начала развиваться в организме, необходимо носительство дефектного гена обоими родителями. При этом чаще всего у них нет проявлений заболевания, а обоюдное носительство не всегда дает появление недуга у совместного ребенка. Доказано, что каждый 40 человек может содержать в своем генетическом материале мутации определенного вида хромосом, обуславливающие носительство дефектного гена.
Проявления заболевания
Выделяют несколько форм развития патологии, которые отличаются возрастом возникновения первых симптомов, тяжестью течения, продолжительностью жизни.
Чем позже развивается патология, тем лучше прогноз для жизни и поддержания навыков самообслуживания.
В любом случае больные становятся инвалидами, требующими дополнительный уход и специальных средств передвижения (коляски, ходунки). Часто пациенты прикованы к кровати, их состояние осложняется застойными процессами в легких, которые могут привести к летальному исходу.

Спинальная мышечная атрофия тип 1 может быть заподозрена еще во внутриутробном периоде при слабом шевелении ребенка во второй половине беременности. На протяжении полугода после рождения отмечают вялость, низкую активность новорожденного. При обследовании не обнаруживают сухожильные рефлексы (ахиллов, коленный).
Мышцы верхних и нижних конечностей, туловища и головы не развиваются. Слабость и атрофия мышечных волокон приводит к неспособности детей полноценно сидеть, ползать, вставать на ноги. По мере роста скелета возникает деформация костей. Например, при попытке новорожденного сидеть позвоночник не поддерживается мышечным каркасом спины, что приводит к развитию кифоза.
Нередко выявляют непроизвольное подергивание мышц, дрожание пальцев вытянутых рук. Мышечная атрофия может визуально сглаживаться чрезмерным развитием подкожно-жировой клетчатки. Из-за недоразвития межреберных мышц грудная клетка приобретает уплощенную форму. Страдает функция глотания, сосания, дыхания, в результате чего развиваются тяжелые осложнения: аспирационная и застойная пневмония, истощение, дыхательная недостаточность.
Развитие умственной сферы проходит удовлетворительно, так как отделы головного мозга не подвергаются патологическим изменениям.
Эта форма заболевания считается неблагоприятной для жизни, такие дети обычно погибают в течение первых лет после рождения.
Атрофия спинного мозга в случае 2 типа болезни проявляется в конце первого года жизни, обычно в 7−18 месяцев ребенка. Признаки патологии выражены не резко и развиваются постепенно. По мере взросления новорожденных отмечают слабую двигательную активность. Дети предпочитают лежать, неохотно перемещаются в пространстве, значительно отстают в физическом развитии от своих сверстников.
Мышцы тела и конечностей подвержены непроизвольным сокращениям, отмечают подергивание языка. Дети способны сидеть, вставать на ноги, иногда медленно передвигаться с посторонней помощью. Прием пищи и дыхание обычно не затруднено, сухожильные рефлексы развиты слабо. Продолжительность жизни таких больных снижают частые респираторные инфекции и тяжелые застойные воспаления легких в результате вялой двигательной активности.
Первые симптомы недуга появляются в конце второго года жизни ребенка, иногда развитие мышечной атрофии начинается в период полового созревания. Заболевание характеризуется медленным прогрессированием. Сначала атрофируются верхние мышечные группы ног, затем патологический процесс поражает руки, туловище, шею. 
Больные, страдающие 3 типом болезни, способны передвигаться: вставать, ходить, подниматься по лестнице. Характерным признаком заболевания считается гипертрофия ягодичных и икроножных мышц. Однако по мере нарастания дистрофических изменений в передних отделах спинного мозга сухожильные рефлексы угасают, что свидетельствует о необратимых изменениях мышечной ткани.
Со временем двигательная активность снижается, что заставляет больных пользоваться инвалидными колясками. Длительно сохраняется способность к самообслуживанию, тазовые функции развиты хорошо (мочеиспускание, дефекация). Чувствительная иннервация при заболевании не страдает.
Терапевтическая тактика
Лечение спинальной мышечной атрофии имеет поддерживающий характер и проводится в течение всей жизни больного.
Выздоровления достичь невозможно, а лишь замедлить развитие патологии в нервной и мышечной ткани.
Терапевтическая тактика включает комплекс мер, направленных на сохранении двигательной активности.
- Лекарственные средства, нормализующие проведение нервного импульса — прозерин, нивалин.
- Ноотропные препараты, улучшающие кровообращение нервной ткани — пирацетам, ноотропил.
- Средства, стимулирующие кровоток и обменные реакции в мышечной ткани — актовегин, оротат калия.
- Витаминные препараты, обладающие антиоксидантным действием — токоферол, витамины группы В, глутаминовая кислота.
- Физиопроцедуры, усиливающие кровообращения пораженных конечностей — парафин, УВЧ, электрофорез с прозерином.
- Массаж — стимулирует мышечную активность, способствует выведению продуктов обмена.
- Лечебная гимнастика — атрофия мышц спины, рук, ног после занятий замедляется.
- Ортопедическая помощь — использование специальных приспособлений, поддерживающих двигательную активность грудной клетки, верхних и нижних конечностей.
В терминальных стадиях заболевания появляются дыхательные расстройства, которые требуют перевода пациентов на искусственную вентиляцию легких.
Спинальная мышечная атрофия считается тяжелым генетическим заболеванием с постоянным прогрессированием патологического процесса и неблагоприятным прогнозом.
В последние годы ученые всего мира ведут научные разработки по синтезированию лекарственного препарата, способного замещать недостающий нейронный белок. Эта единственная надежда больных на выздоровление и значительное улучшение качества жизни.
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
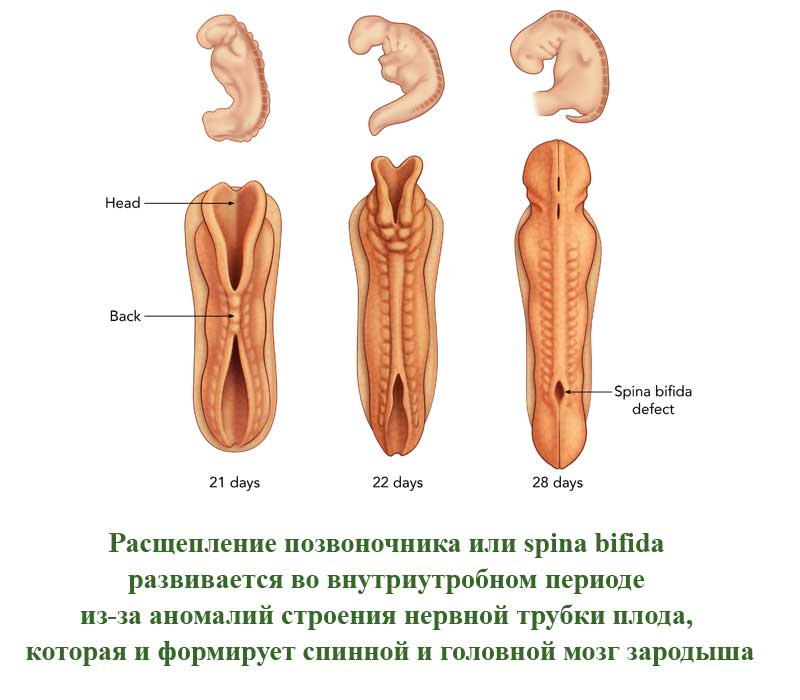
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
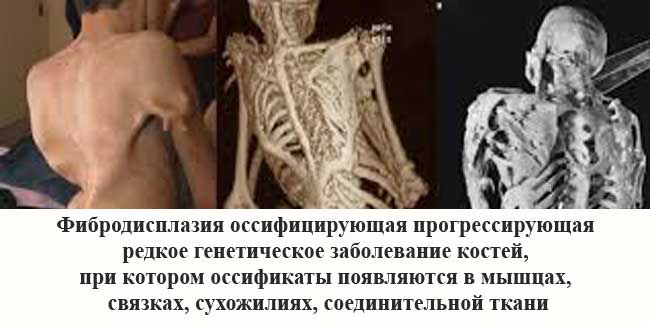
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.
Нервно-мышечными заболеваниями (НМЗ) является группа патологий, которые передаются на генетическом уровне от родителей детям. Нарушаются мышечные функции, снижается двигательная активность. Появляются характерные клинические симптомы.
Патологические процессы развиваются на фоне нарушений функций нервно-мышечных соединений, при поражении мышц и спинномозговых нейронов, нервов. Правильно подобранная терапия не поможет полностью вылечить человека, но позволит улучшить качество его жизни.
Этиология и неврология
Нервно-мышечные заболевания нарушают нормальную синаптическую передачу импульсов с нервных окончаний к мышечным волокнам. В основе каждого типа патологических изменений лежат аутоиммунные процессы.
Большая группа заболеваний характеризуется не только поражением мышечной ткани, но и периферических нервов, передних рогов спинного мозга. Среди часто диагностируемых патологий выделяют миопатию, миотонию, миастению.
Классификация
Нервно-мышечные заболевания различают по следующим видам:
Описание
Нервно-мышечные заболевания передаются по наследству, чаще появляются у людей, в семье которых были родственники с таким диагнозом.
Приобретенные патологические процессы развиваются в результате гормональных или метаболических нарушений в организме человека. Наблюдается сбой в функционировании иммунной системы. Она вырабатывает клетки, которые атакуют свой организм. Аутоиммунные заболевания приводят к появлению слабости в мышцах.
Нервно-мышечные патологии, сопровождающиеся дистрофическими процессами, поражают следующие области тела человека:
- мышцы;
- нервно-мышечные окончания;
- двигательные нейроны;
- периферические нервы.
При миопатии у человека высоки шансы стать инвалидом в результате утраты подвижности. Все виды нервно-мышечных заболеваний без своевременной терапии влекут за собой последствия. Это может быть не только инвалидность, но и смерть человека.
Стадии и степени
Нервно-мышечные заболевания протекают по стадиям. Определить этап развития патологических процессов поможет врач невролог при помощи медицинской диагностики.
| Название | Описание |
| I стадия | Двигательные нарушения слабо выраженные. |
| II стадия | У больного присутствуют ярко выраженные клинические признаки и наблюдаются серьезные двигательные изменения. |
| III стадия | Пациент не может самостоятельно передвигаться. |
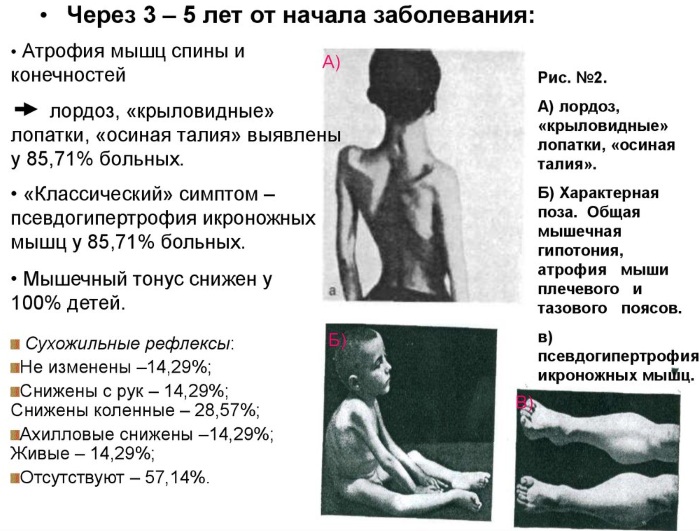
Нервно-мышечные заболевания миопатия Дюшенна на 2 этапе
Клиническая картина зависит от скорости развития патологических процессов и степени тяжести заболевания. Установить точный диагноз поможет врач невролог.
Симптомы
Основной признак нервно-мышечных заболеваний – это слабость мускулатуры. Клиническая картина зависит от области поражения (плечевой пояс, бедра, таз, нижние конечности).
В большинстве случаев у пациентов диагностируют следующие симптомы:
- снижается мышечный объем;
- наблюдаются болезненные спазмы;
- непроизвольно сокращаются мышцы;
- пораженные ткани немеют;
- снижаются сухожильные рефлексы;
- больной ощущает покалывание;
- двоится в глазах (диплопия);
- нарушаются глотательные и дыхательные рефлексы.
При нервно-мышечных заболеваниях опускаются веки, мышечная слабость проявляется симметрично и постепенно прогрессирует. В большинстве случаев при развивающейся мышечной дистрофии слабость возникает в области тазового и плечевого пояса. То же самое касается проксимальных отделов конечностей.
Иногда невральная амиотрофия сопровождается парестезией, нарушением глубокой или поверхностной чувствительности. Клинические признаки нервно-мышечных заболеваний проявляются постепенно. По мере прогрессирования патологических процессов человек теряет способности самостоятельно обслуживать себя. То же самое касается передвижения.
Причины появления
Нервно-мышечные заболевания в большинстве случаев возникают по причине аутоиммунных патологий.
Провоцирующим фактором также являются следующие обстоятельства:
- наследственный фактор;
- поражение периферических нервов и мотонейронов спинного мозга;
- сбои в функционировании нервно-мышечных соединений;
- отравление организма различными веществами;
- врожденный сбой метаболизма;
- патологические изменения в мышцах.
Нервно-мышечные заболевания также развиваются на фоне нарушений работы двигательного нейрона в области ствола головного мозга.
Определить причину и поставить точный диагноз поможет врач невролог. Учитывая состояние пациента, степень развития патологических процессов и индивидуальные особенности человеческого организма, специалист подберет эффективное лечение.
Диагностика
Медицинское обследование позволит врачу установить точный диагноз. Тестирование специалист назначает пациенту, учитывая его жалобы и симптоматику.
Для диагностики нервно-мышечных заболеваний назначаются следующие методы обследования:
Описание

Исследованием состояния сердечной мышцы занимается кардиолог. Специалист назначает не только кардиограмму, но и ультразвуковое исследование (УЗИ) сердца.
Когда необходимо обратиться к врачу
К врачу необходимо обратиться сразу, при появлении первых признаков нарушений в работе мышц. Но если в семье есть родственники с нервно-мышечными заболеваниями, необходимо пройти полное медицинское обследование и понять, насколько высока вероятность появления патологических процессов по наследственной линии.
Диагностикой и лечением занимается врач невролог. Специалист проведет осмотр и подберет максимально информативные методы исследования.
Профилактика
Нервно-мышечные заболевания в большинстве случаев развиваются по причине наследственного фактора. Предупредить патологические изменения невозможно. Женщине во время планирования беременности рекомендуется проходить медицинские обследования, особенно если в семье есть родственники с таким диагнозом.
Диагностические мероприятия также назначаются в период вынашивания малыша на ранних сроках. При высокой вероятности развития нервно-мышечных заболеваний специальная медицинская комиссия советует будущей матери прервать беременность.
Методы лечения
Терапия нервно-мышечных заболеваний осуществляется комплексными методами. Пациентам назначают медицинские препараты, лечебную физкультуру. При отсутствии серьезных противопоказаний, можно использовать рецепты знахарей и целителей. Основная цель терапии – это поддержать мышечные силы и замедлить атрофирующие процессы.
Медикаменты подбирает врач невролог, учитывая результаты медицинской диагностики, степень развития патологических процессов и индивидуальные особенности организма человека.
Самостоятельно не рекомендуется принимать лекарства, поскольку многие препараты вызывают побочные эффекты.
При нервно-мышечных заболеваниях врач назначает следующие медикаменты:
Применение
Лекарства позволяют устранить дефицит энергии и белка, положительно влияют на вещественные обмены в мышечных тканях. Дополнительно назначаются витаминные комплексы.
Нервно-мышечные заболевания можно лечить рецептами знахарей и целителей, но в качестве вспомогательной терапии. Народные средства улучшают качество жизни пациента и общее его состояние. Используемые средства следует обсуждать с лечащим врачом неврологом.
| Название | Рецепт | Применение |
| Овес | Зерна хорошо промыть и залить водой (500 мл). Полученную массу ставят на огонь, доводят до кипения и греют 30 мин. Дальше оставляют на 2 часа, процеживают и принимают по схеме. | Готовое средство рекомендуется принимать внутрь перед едой 4 раза в сутки. Курс терапии продолжается 3 месяца. Затем необходимо сделать перерыв на 30 дней и продолжить терапию. |
| Репчатый лук | Продукт очистить и смешать 200 г с сахаром (200 г), добавить воды (0,5 л). Полученную массу поставить на медленный огонь и греть 1,5 часа. Остудить и добавить 2 ст.л. натурального меда. | Готовое средство рекомендуется принимать по 2 ч.л. 3 раза в сутки. |
| Чеснок | Очистить и измельчить 3 головки чеснока. Добавить 4 лимона, предварительно измельченные. Все компоненты залить медом (1 л) и льняным маслом (200 г). | Полученное средство следует принимать по 1 ч.л. 3 раза в день. |
При нервно-мышечных заболеваниях полезно проводить контрастные ванны для нижних конечностей. После водных процедур ноги рекомендуется укутывать теплым одеялом.
Комплексная терапия нервно-мышечных заболеваний позволяет замедлить их развитие, продлевает период ремиссии и улучшает качество жизни пациента.
Вместе с традиционным и народным лечением больным назначаются следующие методы терапии:

| Название | Описание |
| Физиотерапевтические процедуры | Лечение улучшает проводимость нервных импульсов в мышечных тканях, способствует их питанию. Усиливается кровообращение и вещественный обмен. |
| Массаж | Точечное воздействие помогает повысить тонус мышц. Для достижения лечебного эффекта необходимо провести несколько сеансов на протяжении года. |
| Лечебная физкультура | Гимнастика проводится в специализированном комплексе под наблюдением специалиста. |
Сохранить самостоятельное передвижение пациента позволяют специальные ортопедические приспособления. Лечебная физкультура в виде активных и пассивных движений улучшает состояние больного.
Упражнения следует выполнять регулярно, соблюдая умеренные нагрузки. При нервно-мышечных заболеваниях также рекомендуется плавать. В воде легче выполнять физические упражнения без нагрузки на позвоночник.
Возможные осложнения
Негативные последствия патологических процессов появляются в результате поражения различных внутренних органов и систем организма человека:
Описание
Прогрессирующие патологические процессы также могут спровоцировать искривление позвоночника (кифоз, сколиоз). Больным необходимо носить специальные корсеты. В тяжелых ситуациях или на запущенных стадиях развития нервно-мышечных заболеваний пациенту показано оперативное вмешательство. Решение принимает врач невролог, учитывая состояние человека и индивидуальные особенности его организма.
При нервно-мышечных заболеваниях нарушается двигательная функция, слабеют мышцы. Симптомы постепенно усиливаются на фоне прогрессирующих дистрофических процессов.
Пациенту необходимо пройти полное обследование для постановки диагноза и специально подобранное лечение. Лекарства, средства народной медицины, физиотерапевтические процедуры помогут лишь облегчить жизнь пациенту, но полностью избавить от генетической патологии не смогут.
Оформление статьи: Владимир Великий
Видео о нервно-мышечных заболеваниях
Телесеминар о нервно-мышечных заболеваниях:
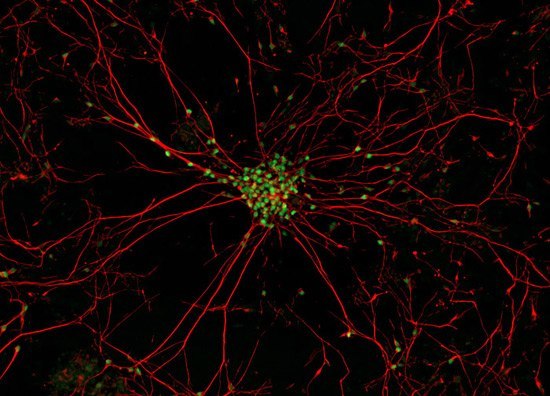
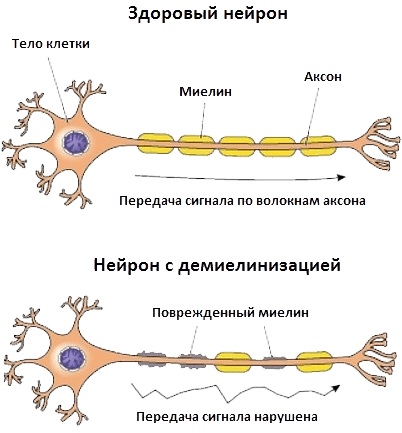
Настоящее фото мотонейрона (двигательного нейрона в передних рогах спинного мозга) - закупорка межсинаптических щелей (зелёные точки) - блокада передачи импульсов через медиаторы (вещества для передачи импульсов) в синапсы (места соединения) отростков нервных клеток.
В центре - тело мотонейрона.
Красные линии -длинные отростки мотонейрона - аксоны и короткие -дендриты.
Причина появления блокады передачи импульсов в межсинаптических щелях учёными мира не найдена. Предположительно - мутация гена,кодирующего фермент передачи этих импульсов через нейромедиаторы - супероксиддисмутазы. (СОД).
Предрасположенность к мутациям гена может иметь наследственный характер по аутосомно-рецессивному типу.
Эндемические (массовые вспышки) случаи этого заболевания зафиксированы у групп военных, живущих в островах на тихом океане. Чаще болеют мужчины от 40 до 60 лет.Следовательно не исключается инфекционная причина развития заболевания.
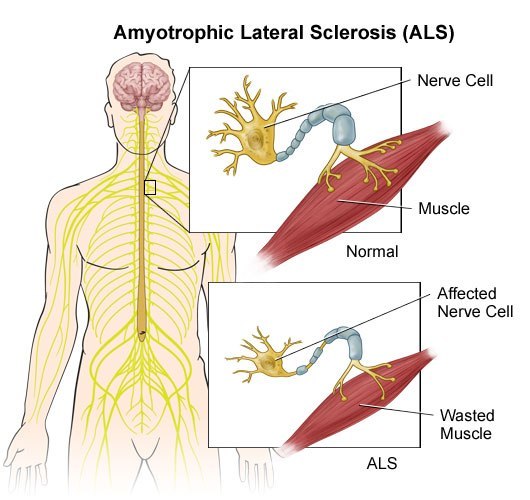
Боковой амиотрофический склероз. (БАС)..
Показано истончение нервных волокон в случае БАС и нарушение иннервации (передачи нервных импульсов) к мышцам. Как следствие - уменьшение работы мышцы и её последующая атрофия. (уменьшение размеров, обратное развитие.)
Блокада передачи нервных импульсов к мышцам (как к поперечно-полосатым которыми мы управляем сами своей волей так и к к гладким, работающим, независимо от нашего сознания, усилий и воли) пищеварительной и дыхательной системы ведёт к смерти из-за невозможности совершать эти жизненно важные моменты работы мускулатуры.
Две статьи из медицинских источников:
1) Теория аксостаза бокового амиотрофического склероза. Аксональная теория бокового амиотрофического склероза
Теория аксостаза основана на анализе патологических процессов, происходящих в аксональном транспорте мотонейронов [Chou S., 1992]. Наибольшими нейронами организма являются двигательные мотонейроны передних рогов спинного мозга и пирамвды Беца. Они должны поддерживать интеграцию дендритов, часто протяженностью более 1 см, и аксон, достигающий 100 см. В аксоне имеются непрерывные потоки, через которые клеточное тело направляет структурные и функциональные белки на периферию и получает обратные сигналы. Ортоградный транспорт бывает 2 видов: а) быстрый — 400 мм в день, идущий в обоих направлениях и транспортирующий связанные с мембраной белки и гликопротеиды, б) медленный — несколько миллиметров в день, транспортирующий сети микрофиламентов, микротрубочек, нейрофиламентов, как компонент "а" (0,1—2 мм в день), а также большой комплекс растворимых белков, как компонент "б" (2—4 мм в день). Ретроградный аксональный транспорт несет эндогенные (аминокислоты, фактор роста нервов) и экзогенные (токсин столбняка, вирус полиомиелита, простого герпеса, бешенства, лектин пероксидазы хрена и др.) субстанции от терминальных аксонов к клеточному телу со скоростью свыше 75 мм в день. Морфологические исследования аксонального транспорта в биоптатах двигательных веточек периферических нервов больных боковым амиотрофическим склерозом выявили уменьшение скорости ретроградного аксонального транспорта и, следовательно, связи терминального аксона с перикарионом [Bieuer A. et al., 1987]. В межреберных нервах больных АБС еще до развития признаков нейрональной дегенерации появляются изменения белков микротрубочек [Binet S. et al., 1988].
Улыраструктурные исследования проксимального аксона и аксонального бугорка мотонейронов переднего рога спинного мозга больных, умерших от бокового амиотрофического склероза [Sasaki S. et al., 1996], показали нарушение быстрого аксонального транспорта. Гладкий эндо-плазматический ретикулум теряет структуру: происходит скопление митохондрий, лизосом, Леви-подобных телец, эозино-фильных и гиалиновых включений, липофусциновых гранул, особенно в аксональном бугорке. Присутствие этих необычных структур является отражением дисфункции аксонального транспорта. Применительно к возможной этиологии АБС еще ранее выдвинута концепция "аксостаза" [Chou S., 1992]. Ней-ротоксические факторы путем ретроградного транспорта избирательно поражают нейрон, создавая феномен "суицидцального транспорта". Ухудшение медленного транспорта в аксоне сопровождается скоплением нейрофиламентов, набуханием проксимального аксона и последующей дистальнои аксональной атрофией, а также вторичной демиелинизацией, характерной для центральной дистальнои аксонопатии или "ретроградного умирания" — "dying back". Определенную значимость в развитии ранних морфологических изменений мотонейронов при АБС имеет теория аутоиммунитета [Smith R. et al., 1996], основанная на появлении антител к зарядам входа кальциевых каналов. Пассивный перенос фракций, содержащих иммуноглобулин, мышам вызывает изменения нервно-мышечных соединений, сходные с таковыми при спорадическом АБС. У животных эти изменения отражают расстройства внутриклеточного Са2+ гомеостаза, и раннее повреждение пластинчатого комплекса в мотонейронах в форме набухания и фрагментации. Иммуноглобулины от больных спорадическим боковым амиотрофическим склерозом вызывают зависимый от Са2+ апоптоз клеток вследствие оксидативных повреждений. Апоптоз, обусловленный иммуноглобулином от указанных больных, регулируется присутствием связанных белков, которые могут модулировать избирательную ранимость нейронов при спорадическом АБС.
2) Боковой амиотрофический склероз
Несмотря на более чем 100-летнее изучение, боковой амиотрофический склероз (БАС) остается фатальным заболеванием центральной нервной системы. Заболевание характеризуется неуклонно прогрессирующим течением с избирательным поражением верхнего и нижнего мотонейронов, что приводит к развитию амиотрофий, параличей и спастичности. До настоящего времени вопросы этиологии и патогенеза остаются невыясненными, в связи с чем не разработаны специфические методы диагностики и лечения этого заболевания. Рядом авторов отмечено повышение частоты встречаемости заболевания среди лиц молодого возраста (до 40 лет).
МКБ-10 G12.2 Болезнь двигательного неврона
ЭПИДЕМИОЛОГИЯ
Боковой амиотрофический склероз дебютирует в возрасте 40 – 60 лет. Средний возраст начала заболевания 56 лет. БАС - болезнь взрослых, и не наблюдается у лиц моложе 16 лет. Несколько чаще заболевают мужчины (отношение мужчины-женщины 1,6-3.0: 1).
БАС является спорадическим заболеванием и встречается с частотой 1,5 – 5 случая на 100 000 населения.
В 90% случаев БАС носит спорадический, а в 10% - семейный или наследственный характер как с аутосомно-доминантным (преимущественно), так и с аутосомно-рецессивным типами наследования. Клинические и патоморфологические характеристики семейного и спорадического БАС практически идентичны.
В настоящее время возраст является основным фактором риска при БАС, что подтверждается нарастанием заболеваемости после 55 лет, и в этой возрастной группе уже не наблюдается различий между мужчинами и женщинами. Несмотря на достоверную связь БАС с возрастом, старение является только одним из предрасполагающих факторов развития патологического процесса. Вариабельность заболевания как в различных возрастных группах, так и среди лиц одного возраста предполагает существование определённых факторов риска: дефицит, или наоборот, наличие определённых нейропротективных факторов, к которым в настоящее время относят: нейростероиды или половые гормоны; нейротрофические факторы; антиоксиданты.
Некоторые исследователи отмечают особо благоприятное течение заболевания у молодых женщин, что подтверждает несомненную роль половых гормонов, в особенности эстрадиола и прогестина, в патогенезе бокового амиотрофического склероза. Подтверждением этому являются: большая частота встречаемости БАС у мужчин до 55 лет (при этом у них отмечается более раннее начало и быстрое прогрессирование заболевания по сравнению с женщинами); с наступлением менопаузы женщины болеют также часто, как и мужчины; единичные случаи заболевания боковым амиотрофическим склерозом во время беременности. К настоящему времени существуют единичные работы по изучению гормонального статуса больных с боковым амиотрофическим склерозом, и ни одной, посвящённой определению концентраций гормонов у молодых пациентов.
Этиология заболевания не ясна. Обсуждается роль вирусов, иммунологических и метаболических нарушений.
В развитии семейной формы БАС показана роль мутации в гене супероксиддисмутазы-1 (Cu/Zn-супероксиддисмутазу, SOD1), 21q22-1 хромосома, выявлен также БАС, связанный с 2q33-q35 хромосомой.
Синдромы, клинически не отличимые от классического БАС, могут возникать в результате:
•опухоли большого затылочного отверстия
•спондилез шейного отдела позвоночника
•артериовенозная аномалия спинного мозга
•бактериальные - столбняк, болезнь Лайма
•вирусные - полиомиелит, опоясывающий лишай
Интоксикации, физические агенты:
•токсины - свинец, алюминий, другие металлы.
Читайте также: