
Генетическое заболевание костей лица
Синдром Тричера Коллинза ( челюстно-лицевой дизостоз) — аутосомно-доминантное заболевание, характеризующееся черепно-лицевой деформацией. Описал его английский офтальмолог Эдвард Тричер Коллинз в 1900 году.
Данная патология встречается очень редко. Ее частота – 1 случай на 50 000 новорожденных. Кроме того, этот синдром отличается благоприятным прогностическим критерием для последующей жизни: у этих больных не нарушено умственное развитие, и при незначительных стадиях развития они могут продолжать ведение активной социальной жизни. Большой угрозой являются тяжелые стадии синдрома Тричера Коллинза — при них у ребенка практически полностью нет лица, и заболевание является тяжелейшим уродством, которое полностью отрезает ребенка от социума.
Причины патологии
Причины синдрома Тричера Коллинза – мутации гена TCOF1 (в локусе хромосомы 5q31.3-33.3), который кодирует ядрышковый фосфопротеин, отвечающий за формирование черепно-лицевой части эмбриона человека. В результате преждевременного уменьшения количества этого белка нарушаются биогенез и функции рРНК. По мнению генетиков исследовательской программы Human Genome, эти процессы приводят к сокращению пролиферации эмбриональных клеток нервного гребня – валика вдоль нервного желоба, который в ходе развития зародыша замыкается в нервную трубку.

Формирование тканей лицевой части черепа происходит благодаря трансформации и дифференциации клеток верхней (головной) части нервного гребня, которые мигрируют вдоль нервной трубки в область первой и второй жаберных дуг зародыша. И дефицит этих клеток вызывает черепно-лицевые деформации. Критический период возникновения аномалий – с 18 по 28 день после оплодотворения. По завершении миграции клеток нервного гребня (на четвертой неделе гестации) образуются практически все рыхлые мезенхимальные ткани в области лица, которые позже (с 5 по 8 недели) дифференцируются в скелетные и соединительные ткани всех частей лица, шеи, гортани, уха (в том числе внутреннего) и будущих зубов.
Симптомы
У каждого человека заболевание проявляется по-разному. Могут наблюдаться один или два симптома, или же полный набор признаков. Но ребенка с синдромом Тричера Коллинза можно узнать сразу. Фото больных детей есть в интернете, часто они похожи друг на друга. У заболевания могут быть такие внешние признаки:

Стадии заболевания
Признаки синдрома Тричера Коллинза могут проявляться, начиная от почти незаметных до очень тяжелых деформаций. В соответствии с этим выделяют три стадии развития заболевания.
- На начальной стадии деформации костей практически незаметны. Дети с такой патологией могут вести нормальную жизнь.
- Вторая стадия характеризуется серьезными аномалиями развития костей черепа. К ним присоединяются патологии слуха, зубов, сложности с дыханием или принятием пищи.
- Третья самая тяжелая стадия заболевания характеризуется почти полным отсутствием лица у ребенка.
Лечение
В случае, когда заболевание диагностировано еще во время внутриутробного развития, женщине рекомендуют сделать аборт. В противном случае, сразу после рождения ребенку потребуется квалифицированное комплексное лечение по устранению дефектов, насколько это возможно. Терапия заключается в следующих действиях:
- Хирургическое вмешательство, в ходе которого устраняются дефекты лица. Обычно необходимо несколько операций, чтобы уменьшить проявления болезни. Все зависит от сложности случая.
- Стоматологические процедуры. Обычно у ребенка диагностируют не только проблемы с зубами, но и не правильный прикус, которые с помощью дантистов можно корректировать.
- Чтобы улучшить костнопроводимость звуков, потребуется установить слуховые аппараты.
Если у ребенка наблюдаются проблемы с дыханием и питанием, то потребуются операции по решению этих проблем (трахеотомия и гастростомия). Для дальнейшей адаптации ребенка потребуется работа с логопедами и сурдопедагогами.
Профилактика
В большинстве случаев костные деформации при синдроме Тричера Коллинза не несут угрозы для жизни ребенка. Дети с данной патологией не отстают в интеллектуальном развитии при условии своевременной коррекции слуховых нарушений. Из-за внешних дефектов они могут испытывать значительные трудности с социальной адаптацией, поэтому им требуется психологическая помощь.
Синдром Тричера Коллинза развивается вследствие генетических мутаций, по этой причине его невозможно предупредить. Парам с негативным семейным анамнезом при планировании беременности рекомендуется проконсультироваться с генетиком.
Как с этим живут?
Синдром Тричера Коллинза – это пожизненный диагноз. Страдающие от него дети обречены на осложнения, которые порой невозможно исправить даже повторными операциями. Из-за грубых пороков развития органов слуха пациенты страдают тугоухостью. Без слухопротезирования они обречены на всю жизнь.
Большие проблемы создают пороки развития ротового аппарата. Деформация костей челюсти, зубного ряда, редуцированные слюнные железы делают прием пищи сложной и порой неосуществимой самостоятельно процедурой.
В большинстве случаев синдром Тричера Коллинза затрагивает внешние деформации. Заболевание не откладывает отпечаток на умственное развитие ребенка. Возможные логопедические проблемы, связанные с дефектами ротовой полости, потенциально устранимы. Такие дети имеют шанс на адаптацию в обществе.
Несмотря на этот тяжелый пожизненный диагноз, многие люди с синдромом Тричера Коллинза смирились с ним и ведут полноценный образ жизни.
В медицинской практике данная патология встречается крайне редко. При этом синдром Тричера-Коллинза – это врожденное заболевание, причины которого обусловлены тем, что измененный вследствие мутационных процессов ген родителей наследуется ребенком, организм которого еще на этапе эмбриогенеза начинает испытывать тяжелые последствия этого состояния. Узнайте о проявлениях этого недуга, а также о современных способах его диагностики и лечения.
Что такое синдром Тричера-Коллинза
Указанное патологическое состояние является сугубо генетически обусловленным недугом, который характеризуется врожденной деформацией костей черепа, или челюстно-лицевым дизостозом. В медицинской среде болезнь Тричера-Коллинза имеет и другое название – синдром Франческетти. Заболевание, как правило, наследуется от родителей со спонтанными мутациями генов tcof1.

Симптомы
Синдром Тричера характеризуется полиморфностью клинических проявлений. При этом первые признаки заболевания возникают уже на этапе внутриутробного развития плода, поэтому новорожденный рождается со всеми симптомами аномалии строения черепа. Основным симптомом патологии у больных детей выступают множественные дефекты лицевых костей, что заметно даже при мимолетном взгляде на фото страдающих этим недугом. Одним из самых ярких проявлений синдрома является нарушение нормальной формы глазной щели. Среди прочих симптомов болезни Тричера-Коллинза стоит выделить:
- нарушение развития костной структуры скул, нижней челюсти;
- дефект мягких тканей ротовой полости;
- отсутствие ушных раковин;
- колобомы век;
- впалый подбородок;
- нарушение слуха;
- расщепление верхнего неба;
- нарушение прикуса.
- Как приготовить камбалу в духовке
- Куриные отбивные: рецепты с фото
- Клизма для похудения - домашние рецепты. Очистительная клизма - отзывы о результатах похудения
Причины заболевания
Синдром Тричера – генетическое заболевание, на возникновение которого в большинстве случаев не влияют никакие внешние или внутренние факторы. Можно сказать, что патология изначально заложена в аминокислотный код будущего ребенка и начинает проявляться задолго до его рождения. Научно доказано, что спонтанные изменения в структуре ДНК (генные мутации) у лиц, имеющих синдром, возникают в 5 хромосоме. Последняя является самой длинной нуклеотидной структурой в геноме человека и отвечает за производство материала для скелета плода.
Происходят мутации по причине сбоя внутриклеточного синтеза белка. В результате чего развивается синдром гаплонедостаточности. Последний характеризуется нехваткой белка, необходимого для правильного развития лицевой части черепа. При всем этом следует знать, что болезнь Тричера-Коллинза имеет аутосомно-доминантный, реже – аутосомно-рецессивный характер. Дефект генов наследуется детьми от больных родителей только в 40% случаев, тогда как остальное 60% возникают вследствие новых мутаций, которые нередко вызывают следующие тератогенные факторы:
- этанол и его производные;
- цитомегаловирус;
- радиоактивное излучение;
- токсоплазмоз;
- прием противосудорожных и психотропных препаратов, лекарств с ретиноевой кислотой.
Стадии развития болезни
Болезнь Тричера-Коллинза имеет три стадии. На начальном этапе его развития наблюдается незначительная гипоплазия лицевых костей. Вторая стадия характеризуется деформацией и недоразвитостью слуховых проходов, маленькой нижней челюстью, аномалиями глазной щели, что прослеживается практически на всех фото пациентов с синдромом. Тяжелые формы патологии сопровождаются практически полным отсутствием лица. При этом проявляются признаки редкого заболевания постепенно и с возрастом (как это видно при ретроспективном анализе фото пациентов) проблема усугубляется.

Осложнения
Одним из самых серьезных последствий синдрома Тричера принято считать недоразвитие ротового аппарата. Значительная деформация зубов, челюстей и отсутствие слюнных желез приводят к отсутствию у больных способности принимать самостоятельно пищу. Кроме того, врожденная аномалия может провоцировать появление заболеваний дыхательной системы по причине больших размеров языка и зарастания носовых проходов.
- Как приготовить маринад для курицы в духовке
- Белковая пища список продуктов
- Пневмосклероз легких - что это такое, как лечить
Диагностика
Пренатальное исследование челюстно-лицевых аномалий проводится на 10-11 неделе беременности при помощи биопсии ворсин хориона. Процедура в достаточной степени опасная, поэтому в дородовой диагностике синдрома Тричера врачи предпочитают использовать УЗИ. Кроме того, у членов семьи берутся анализы крови. На 16-17 неделе беременности проводится процедура трансабдоминального амниоцентеза. Спустя некоторое время назначается фетоскопия и берется кровь из плодовых плацентарных сосудов.
Постнатальная диагностика проводится на основании имеющихся клинических проявлений. При полной экспрессивности синдрома Тричера вопросов, как правило, не возникает, чего нельзя сказать, когда обнаруживаются незначительные признаки данной патологии. В этом случае проводится комплексная диагностика состояния, включающая следующие исследования:
- оценку и мониторинг эффективности кормления;
- аудиологическое тестирование слуха;
- рентгеноскопию черепно-лицевой дисморфологии;
- пантомографию;
- КТ или МРТ головного мозга.
Аналогичные методы исследования применяются, когда необходимо провести дифференциальную диагностику для того, чтобы распознать неярко выраженные проявления болезни Тричера-Коллинза и отличить их от признаков других патологических состояний. Так, в большинстве случаев специалисты назначают дополнительные инструментальные исследования для дифференциации указанного недуга с синдромами Гольденхара (гемифациальной микросомии), Нагера.
Лечение синдрома Тричера-Коллинза
Сегодня не существует терапевтических методов, позволяющих помочь людям с деформацией структур лицевого черепа. Лечение пациентов носит исключительно паллиативный характер. Тяжелые формы синдрома являются показанием для операции. С целью коррекции слуха тем, кто страдает редкой аномалией ушных раковин, рекомендуется ношение слухового аппарата. При всем этом нельзя забывать о психологической помощи больным синдромом Тричера. Поддержка со стороны членов семьи, друзей играют важную роль в последующей нормальной социальной адаптации лиц с черепно-лицевым дизостозом.

Синдром Тричера Коллинза (СТК) — генетически детерминированная патология, обусловленная мутацией гена 5 хромосомы и проявляющаяся внешними уродствами вплоть до обезображивания лица. Дефектное формирование костей черепа происходит в период внутриутробного развития плода. Недоразвитые челюстные, скуловые, лобные и другие кости придают характерный внешний вид больным детям. Поскольку заболевание является врожденным, такие аномалии обнаруживают сразу после родов. Современные методы диагностики позволяют выявить синдром внутриутробно и предотвратить рождение больного ребенка.
Синдром получил свое название в честь врача-офтальмолога из Англии, который в начале 20 века впервые описал клинические признаки и этиопатогенетические особенности недуга. Патология встречается у 1 из 50 тысяч новорожденных. В настоящее время синдром изучен не полностью, хотя в мире проживает много людей с его различными формами.

мальчик с синдромом Тричера-Коллинза
Заболевание обычно не представляет серьезной опасности для жизни больных и не оказывает влияния на их умственное развитие. В отдельных случаях грубая деформация черепа мешает ребенку самостоятельно дышать и принимать пищу. Ограничивая дыхательные пути больному, недоразвитые лицевые кости становятся причиной опасных для жизни заболеваний.
Этиология и патогенез
Единственным этиопатогенетическим фактором синдрома Тричера Коллинза считается генетическая мутация. Врожденная аномалия структуры пятой хромосомы является причиной недуга. Это самая длинная нуклеотидная структура в геноме человека, отвечающая за производство материала для скелета плода. В организме больного нарушается биогенез и функции рибосомной РНК, происходит сбой внутриклеточного синтеза белка, замедляется процесс деления эмбриональных клеток нервной трубки. Их самоуничтожение приводит к недоразвитию костной ткани и формированию обезображенного лица ребенка на раннем этапе эмбриогенеза. Диагностировать недуг можно уже на втором месяце беременности.

Синдром в 100% случаев наследуется от больной матери или отца по доминантному принципу. Если имеется отягощенный семейный анамнез, то в данной семье с этой патологией обязательно рождаются больные дети. Экспрессивность и пенетрантность гена обуславливают различную степень выраженности дефекта, которая варьируется у разных пациентов от умеренной до крайне тяжелой.
В некоторых случаях синдром не наследуется, а формируется в силу новой мутации генов после зачатия. Рождаются дети с синдромом от абсолютно здоровых родителей. Мутация может произойти под влиянием факторов, оказывающих тератогенное воздействие на плод:
- злоупотребление беременной женщиной алкоголем,
- курение и наркотическая зависимость,
- сильный стресс,
- вирусные и бактериальные инфекции у женщины,
- тяжелые сопутствующие патологические процессы,
- применение определенных лекарств — психотропных и противосудорожных препаратов,
- радиационное облучение.
Симптоматика
Синдром характеризуется полиморфностью клинических проявлений. Больных с СТК можно узнать сразу. Такие дети имеют характерный внешний вид и часто похожи друг на друга.

Клинические признаки синдрома:
Деформация лица и черепа может сочетаться с пороками развития внутренних органов: сердца, позвоночника, слухового анализатора, желез внешней и внутренней секреции, дыхательных путей. У больных детей нарушается адаптация в обществе. Они стесняются окружающих и избегают контактов с ними. Это приводит к формированию комплекса неполноценности и развитию депрессии. При этом интеллект полностью сохраняется: больные адекватно воспринимают информацию и правильно развиваются в моральном и физическом плане.
Клинические признаки синдрома имеют различную степень выраженности: от малозаметных деформаций до тяжелых уродств, при которых полностью стерты черты лица. У больных в запущенных случаях появляются проблемы с жеванием и глотанием, произношением отдельных звуков, зрением и слухом.
Стадии
Стадии синдрома определяются сложностью мутационного процесса и интенсивностью клинических признаков:

СТК тяжелой степени
- Начальная стадия характеризуется практически незаметными изменениями на лице. Больные дети ни чем не отличаются от здоровых и ведут нормальный образ жизни.
- Средняя стадия проявляется всеми перечисленными выше нарушениями. Аномальная деформация лицевых костей достаточно сильная. Возможны трудности с дыханием, принятием пищи, нарушение слуха, проблемы с зубами.
- Тяжелая стадия — полное отсутствие лица, невозможность рассмотреть его черты. Даже пластическая хирургия не может помочь больным.
Осложнения
Тяжелые осложнения и неприятные последствия синдрома Тричера Коллинза:
- тугоухость и полная глухота,
- неспособность принимать пищу,
- удушье,
- полное отсутствие зубов,
- неправильное формирование глазных яблок,
- нарушение глотания,
- аномалия развития зубов, проблемы с жеванием и звукопроизношением,
- гнусавый голос,
- поражение нервной системы и психические расстройства из-за ощущения своей ущербности,
- врожденные пороки сердца и внутренних органов.
Диагностика

Диагностикой и лечением синдрома Тричера Коллинза занимаются педиатры, пластические хирурги, ЛОР-врачи и генетики. Диагностические мероприятия подразделяются на пре- и постнатальные.
- Пренатальное выявление патологии осуществляется во время ультразвукового исследования беременной женщины. Дополнительно проводят хорионбиопсию, амниоцентез и анализ амниотической жидкости, исследование крови из плодовых сосудов плаценты, фетоскопию.
- Постнатальная диагностика основывается на характерных клинических признаках и внешних данных больного. Если симптоматика синдрома выражена незначительно, возникают проблемы с постановкой диагноза. Специалисты должны обратить особое внимание на функцию дыхания и насыщения гемоглобина кислородом, а также оценить эффективность кормления больного ребенка.
Во время молекулярно-генетического исследования находят дефект в 5 хромосоме и обнаруживают мутацию ответственного за заболевание гена. С учетом наследственной предрасположенности и клинической картины ставят диагноз патологии. Генетическое консультирование осложняется переменной экспрессией заболевания.
Дополнительные диагностические методы:
- оценка слуха ребенка — фиксация слуховых потенциалов, аудиометрия, аудиологическое тестирование, томография височных костей;
- оценка органов дыхания связана с риском апноэ во сне;
- рентгеноскопическое или томографическое исследование головы;
- пантомография;
- КТ и МРТ головного мозга.
Лечебные мероприятия
Синдром Тричера Коллинза — неизлечимое заболевание, при котором невозможно устранить первопричину деформаций черепа и лица. Больным показана паллиативная помощь, позволяющая улучшить качество жизни пациентов. Если синдром был диагностирован во время внутриутробного развития, беременным рекомендуют сделать аборт. Когда рождается больной ребенок, ему требуется квалифицированная комплексная терапия. Больным проводят хирургическое и ортодонтическое лечение, улучшающее внешний вид и качество жизни.
- Хирургическое вмешательство проводится с целью исправления внешних недостатков для комфортного нахождения больных в обществе. Операция также позволяет предотвратить смертельный исход при наличии мутаций, затрудняющих дыхание и глотание. Оперативное вмешательство следует проводить как можно раньше, особенно в тяжелых случаях, когда имеется сужение дыхательных путей. Больным выполняют трахеостомию и гастростомию для кормления. Затем переходят к хирургической коррекции неба, удлинению нижней челюсти, эндоскопической полисинусотомии, реконструкции мягких тканей — пластике ушных раковин, коррекции колобомы, супраглоттопластике. Эти операции очень трудоемки и затратны. Ограниченное открывание рта очень трудно поддается коррекции. Специалист по ЛОР-хирургии требуется для лечения патологии среднего и наружного уха.
- Для улучшения слуха используют слуховые аппараты. Слухопротезирование особенно необходимо в тех случаях, когда операции на измененных слуховых косточках дают слабый результат. Чтобы больной ребенок в будущем не отставал в умственном развитии от сверстников, слуховой аппарат следует носить с 3-х месяцев до 3-х лет. В дальнейшем в заушную область устанавливают магнитный имплант.
- Стоматологические процедуры проводят дантисты с целью устранения неправильного прикуса и восстановления зубов.
- Для улучшения речи проводятся логопедические и сурдологические занятия, а для адаптации в обществе — сеансы психотерапии.
- Людям, имеющим проблемы с глотанием пищи или напитков, требуется помощь дефектологов.

Черепно-лицевые дефекты при синдроме Тричера Коллинза полностью устранить невозможно. Лечение патологии длительное, особенно если имеются тяжелые нарушения. Больным потребуется целая серия пластических операций. Весь цикл лечения может растянуться на несколько лет. Если хирургам не удается устранить все уродства, больные живут с ними всю жизнь.
СТК — врожденное нарушение черепно-лицевого развития с характерной двусторонней симметричной ухо-нижнечелюстной дисплазией без аномалий конечностей. Это наследственное заболевание крайне редко встречается в современной медицинской практике.
Профилактика и прогноз
Прогноз при СТК благоприятный в том случае, если он не несет угрозы для жизни ребенка. Своевременная коррекция слуховых нарушений и хирургическое исправление внешних дефектов позволяют легко справиться с социальной адаптацией и обеспечивают нормальное интеллектуальное развитие детей. Они живут, ни чем не отличаясь от своих сверстников, и создают полноценные семьи. Некоторым пациентам требуется помощь психологов, поскольку они видят свое уродство и страдают от этого. Те, кто не смог смириться с подобным недугом, погружаются в депрессию и стараются избегать всяческого общения с окружающими людьми.
СТК — пожизненный диагноз. Заболевание протекает достаточно тяжело и требует высококвалифицированной помощи. Поскольку его причиной является генетическая мутация, предупредить развитие синдрома невозможно. Медико-генетическое консультирование требуется парам с негативным семейным анамнезом. Если данное заболевание не было зафиксировано у близких и дальних родственников, необходимо соблюдать стандартные рекомендации относительно здорового образа жизни во время беременности.
Видео: о самом известном человеке с синдромом Тричера-Коллинза
Синдром Тричера Коллинза был впервые описан британским врачом Томпсоном в 1846 г. На фото пациентов отчетливо прослеживаются одни и те же признаки аномалий лицевых костей. По мере роста ребенка они становятся более выраженными. Наилучший прогноз жизни у тех пациентов, у которых не нарушено развитие внутренних органов.
Общая информация
Синдром Тричера Коллинза, фото которого можно увидеть в этой статье, – это генетическое заболевание, при котором происходит мутация гена 5-й хромосомы, отвечающего за формирование костей лицевой части черепа. Кроме этого, могут быть и другие нарушения.
При данном заболевании прекращается нормальный процесс синтеза и удвоения ДНК. Это происходит на 3-4 неделе после оплодотворения. В результате уже на втором месяце беременности в костях лица формируются участки из соединительной ткани, так как половины генетического материала становится недостаточно для нормального развития организма.
Деление клеток в структурах правой жаберной дуги зародыша нарушается.
Болезнь передается по наследству и может наблюдаться у членов одной семьи в 2-3 поколениях. Она имеет различный характер и степень тяжести, что затрудняет генетическое консультирование (профилактику наследственных болезней). Распространенность составляет 1 случай на 50 тыс. новорожденных.
Измененная копия гена является доминирующей, поэтому риск рождения ребенка с этой патологией высок, даже если болен только один из родителей (около 90% вероятности). В случае, когда данным заболеванием страдают оба родителя, то такая вероятность составляет 100%.
В медицине существуют и другие названия синдрома Тричера Коллинза:
- синдром Франческетти;
- мандибулофациальный дизостоз;
- синдром Томпсона.
Почему появляется заболевание
Несмотря на то, что данная патология наследуется от больных родителей, в 60% случаях происходит первичное развитие новых мутаций, которые вызваны следующими негативными факторами:
- злоупотребление алкогольными напитками;
- воздействие радиации;
- цитомегаловирусная инфекция и токсоплазмоз;
![]()
![]()
- контакт с токсическими химическими веществами;
- прием некоторых лекарственных средств (психотропные, противоэпилептические препараты, а также с содержанием ретиноевой кислоты).

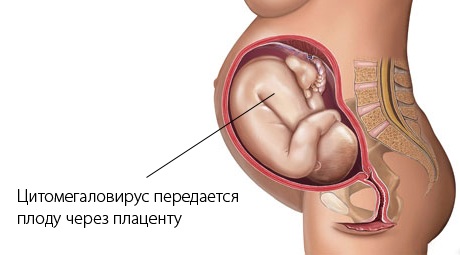
Их влияние на организм женщины до беременности или во время нее может вызвать возникновение дефекта в генах.
Симптомы
Синдром Тричера Коллинза, фото которого наглядно демонстрирует характерные особенности заболевания, обладает следующими основными признаками:
2 последних признака проявляются реже.
Сопутствующие проблемы
Кроме вышеописанных симптомов, могут наблюдаться следующие нарушения:
- аномалии костной системы, в том числе позвоночника;
- отсутствие околоушных слюнных желез;
- врожденные пороки сердца;
- доброкачественные новообразования из хрящевой ткани внутри костей;
- околоушные свищи;
- неопущение яичек в мошонку у мальчиков;
- недоразвитие гайморовых пазух;
- нарушения в кровеносной системе – анемия, нейтропения (снижение концентрации гранулоцитов в крови, приводящее к ухудшению иммунитета), гемолитическая желтуха в результате повышенного разрушения красных кровяных клеток;
- умственная отсталость;
- повышенное оволосение на отдельных участках тела.
У многих новорожденных фиксируется сужение дыхательных путей. Ограниченная возможность для открытия рта и выпадающий язык могут привести к трудностям дыхания и питания в раннем возрасте.
Часто также присутствуют следующие нарушения:
- хроническая дыхательная недостаточность, которая сопровождается одышкой, тахикардией, проявлениями со стороны нервной системы;
- апноэ во время сна – задержка легочной вентиляции более чем на 10 сек., вызывающее состояние гипоксии, а в некоторых случаях – легочную гипертензию с недостаточностью правого желудочка сердца;
![]()
- атрезия хоан – аномальное развитие внутренних носовых ходов, при котором происходит их заполнение соединительной тканью.

Кроме видимых нарушений в наружном аппарате уха, у больных также имеются следующие отклонения:
- двусторонняя тугоухость, возникающая в результате ухудшения передачи звуковых волн от наружного к внутреннему уху (снижение слуха на 50-70 дБ);
- патологическое строение среднего уха, иногда его полное отсутствие;
- полное или частичное отсутствие слуховой трубы;
- пороки развития внутреннего уха (в редких случаях);
- деформация слуховых косточек.
Синдром Тричера Коллинза, для которого характерен специфический разрез глаз, показанный на фото, сопровождается также следующими нарушениями органов зрения:
- недоразвитие глазного яблока;
- врожденная катаракта (помутнение хрусталика, вызывающее снижение зрения);
- кисты по краю роговицы и на конъюнктиве;
- расщепление сосудистой оболочки глаза и зрительного нерва;
- недоразвитость, слабость (или отсутствие) глазодвигательных мышц;
- отсутствие мейбомиевых желез, участвующих в образовании слезной жидкости (чаще на нижнем веке).
На кистях рук и стопах у больных данным синдромом в некоторых случаях отмечается недоразвитие ногтевых пластин.
Диагностика во время беременности
Диагностика заболевания плода в период его вынашивания производится 2 способами: с помощью ультразвукового исследования, на котором выявляются грубые челюстно-лицевые нарушения, и молекулярного исследования образцов биологического материала.
Молекулярно-генетическая диагностика позволяет выявить изменения в ответственном гене путем прямого определения аминокислотной последовательности, которое производится автоматически.
В качестве биологического материала используются:
- образцы ворсин внешней оболочки зародыша, если срок беременности находится в пределах 8-14 недель;
- околоплодные воды, если женщина беременна на сроке 16-21 недель.
Стоимость исследования составляет порядка 30 тыс. руб., срок выполнения около 1 месяца, цена поиска мутации у родственника – 2800-3500 тыс. руб.
Если в семье уже имелись подобные случаи генетических нарушений, то настоятельно рекомендуется сделать диагностику перед подготовкой к беременности (стоимость исследования – 8-10 тыс. руб.). Такое исследование является единственным способом профилактики возникновения данной патологии у ребенка.
Диагностика у новорождённого
Определение заболевания у новорожденного обычно не вызывает затруднений и производится по внешним признакам. Генетический анализ для дополнительного подтверждения диагноза производится по венозной крови, таким же способом, как во время беременности.
Стоимость такого анализа порядка 60 тыс. руб. (вместе с заключением врача-генетика). При выявлении отклонения также рекомендуется сделать исследование для обоих родителей, братьев и сестер.
Дополнительно проводятся следующие обследования:
- определение остроты слуха, методика проведения зависит от возраста ребенка: регистрация вызванных слуховых потенциалов (подача акустических сигналов и регистрация электроэнцефалограммы), речевая аудиометрия (произнесение слов и фраз с различной громкостью), тональная пороговая аудиометрия (проводится с помощью аудиометров, результатом является графическое изображение слухового порога);
- рентгенография черепа;
- компьютерная томография височных костей по достижении возраста 3 лет для подготовки к хирургической операции;
- консультация у профильных специалистов (отоларинголога, окулиста, кардиолога, хирурга и других).
Данное заболевание требует дифференциальной диагностики с такими генетическими патологиями, как:
- Синдром Нагера. Для этой болезни также характерен антимонголоидный разрез глаз, недоразвитие нижней челюсти и патологические изменения в органах слуха и зрения. Кроме этого, у больных детей имеются грубые нарушения в развитии конечностей – укорочение и искривление предплечья, фаланг и локтевых костей, отсутствие пальцев и сращение костей рук.
![]()
- Синдром Гольденхара. Неправильное развитие затрагивает не только лицо и череп, но и нервную, сердечно-сосудистую, мочевыделительную, пищеварительную системы, у многих больных отмечается умственная отсталость. Как правило, данная аномалия имеет односторонний характер (70% больных).

Стадии развития болезни
Синдром Тричера Коллинза, фото которого приведено выше, условно делят на 3 стадии:
- Начальный этап, когда у новорожденного наблюдается незначительное деформирование лицевой части черепа.
- 2-я стадия, на которой происходят нарушение проходимости дыхательных путей, глотания, зрения и слуха.
- 3-я стадия, которая характеризуется грубыми деформациями черепа из-за неравномерного роста костей в течение многих лет.
Первые признаки заболевания можно заметить сразу после рождения ребенка. Их выраженность может быть различной – от едва заметных отклонений до очень тяжелых форм. Анатомические аномалии без хирургического вмешательства постепенно прогрессируют по мере роста и развития ребенка.
Как живут дети с синдромом
В медицинской литературе описано около 250 случаев данного заболевания. И некоторые из них получили мировую огласку.
Так, в 2013 г. вышла передача об американской девочке Джулианне Уэтмор, у которой при рождении была диагностирована самая тяжелая степень этого заболевания в истории медицины. У нее отсутствовало 30-40% костей лица. В прессе ее называли девочкой, родившейся без лица.

Джулианне Уэтмор с синдромом Тричера Коллинза
Несмотря на это, она имеет нормальный уровень интеллекта и является жизнерадостным, общительным ребенком. Ее история стала вирусной через год после рождения в 2003 г. Девочка перенесла несколько десятков пластических операций и теперь ее лицо выглядит гораздо лучше, а ее показательный случай дает надежду другим родителям, столкнувшимся с такой же проблемой.
Семья Джулианны усыновила еще одну девочку с таким же генетическим заболеванием. Первая дочь родилась у них совершенно здоровой, однако, зная о том, что патология передается по наследству, они отказались от идеи рождения 3-го ребенка.
Хирурги надеются, что девочка сможет посещать колледж. Из-за того, что ее челюстные кости сильно деформированы, ей приходится питаться через зонд. Есть сложности и с речью, но она быстро учится языку жестов.
Обучение таких детей чаще всего происходит в специализированных школах для больных с нарушениями слуха. Способность к социальной адаптации во многом зависит от родителей ребенка и выполнения рекомендаций врачей.
Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего. Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их.
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Продолжительность жизни
Синдром Тричера Коллинза, фото которого свидетельствуют об челюстно-лицевых аномалиях, в легких случаях практически не влияет на продолжительность жизни. Гибель пациентов с данным заболеванием наступает при присоединении других патологий, осложняющих его течение. Многие из них доживают до зрелого возраста и даже заводят семьи.
Однако в тяжелых случаях, при множественных аномалиях органов и систем, генетический дефект дает летальный исход еще во внутриутробный период или в первые дни после рождения ребенка.
Об этом свидетельствует высокая частота выкидышей и ранних детских смертей. Врачи отмечают также, что при передаче заболевания к следующему поколению имеется тенденция к увеличению тяжести и осложнений в том случае, если носителем дефектного гена является мать.
Лечебные мероприятия
Как и для большинства генетических заболеваний, специального лечения при данном синдроме не существует. Терапия представляет собой сложную, многопрофильную задачу и зависит от тяжести поражения и осложнений.
Проводятся следующие лечебные мероприятия:
- В случае дыхательной недостаточности – трахеостомия. Эта процедура заключается в создании проходимости в трахее с помощью установки трубки для обеспечения поступления воздуха в дыхательные пути. В легких случаях возможна неинвазивная искусственная вентиляция легких.
- Дистракция (растяжение) нижней челюсти хирургическим путем с помощью специального аппарата. Необходимость проведения такой манипуляции рассматривается врачами в индивидуальном порядке.
- Установка гастростомы – трубки, которая требуется для защиты дыхательных путей от попадания пищи.
- Хирургическая реконструкция лицевой части черепа. Она обычно проводится с 5-6 лет.
При наличии осложнений показаны консультации у соответствующих специалистов и патогенетическое лечение.
Хирургические способы восстановления слуха у таких детей неэффективны. Рекомендуемая тактика реабилитации – использование слуховых аппаратов костной проводимости (или обычных слуховых аппаратов при незначительной деформации ушной раковины).
Их особенностью является то, что звуки передаются во внутреннее ухо по костям черепа. Это не так физиологично, как воздушное звукопроведение, однако при определенном усилении они очень хорошо воспринимаются рецепторами.
При данном заболевании хорошо зарекомендовали себя имплантируемые слуховые аппараты ВАНА (БАХА), которые имеют в своем составе титановую опору, закрепляемую в толще височной кости. Со временем титан срастается с костной тканью, а штифт напрямую передает звуковые колебания в улитку внутреннего уха.
Хирургическая установка имплантата производится в 2 этапа: сначала внедряется титановый штифт, а затем, после его вживления в кость в течение полугода, монтируется опора. Через месяц на нее надевают звуковой процессор.
Такие слуховые аппараты обладают следующими преимуществами:
- у детей отмечается улучшение порогов слышимости в диапазоне громкости обычной речи;
- по сравнению с обычными аппаратами и хирургической реконструкцией эстетические и аудиологические показатели выше;
- происходит спонтанное улучшение речи и постановки голоса (его высоты и интенсивности).
У маленьких детей слухопротезирование проводится с применением эластичной ленты, закрепляемой на голове. Существуют также цифровые аппараты костного проведения звуков, имплантируемые в височную кость (Alpha).
Операция
Аномальное развитие лицевых костей помогает устранить челюстно-лицевая и пластическая хирургия. В сложных случаях требуется проведение целой серии таких операций.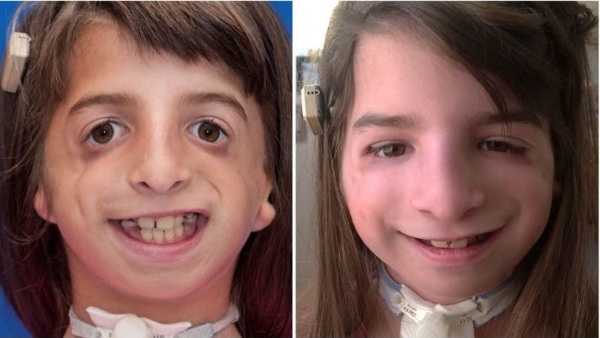
Оперативной корректировке поддаются следующие нарушения:
Этиологического лечения для синдрома Тричера Коллинза не существует. Как показывают фото, степень выраженности этого генетического отклонения может быть различной. Для пациентов применяется терапия, поддерживающая их жизнь на достаточном уровне.
Наиболее распространенным осложнением является ухудшение слуха, для устранения которого используются слуховые аппараты. С целью восстановления анатомических дефектов по возможности проводят пластическую хирургию.
Оформление статьи: Владимир Великий
Видео о синдроме Тричера Коллинза
Девочка с синдромом Тричера Коллинза:
Читайте также: