
Инфекционные заболевания генетическая предрасположенность
Комплексное генетическое исследование, направленное на поиск частых мутаций гена MBL2, который кодирует манноз-связывающий лектин (МСЛ) – белок, участвующий в иммунной защите. Его уровень и активность на 96 % определяются генетическими факторами. В исследование включено 6 генетических маркеров, ассоциированных c геном MBL2 и влияющих на уровень белка в сыворотке крови.
Какой биоматериал можно использовать для исследования?
Буккальный (щечный) эпителий, венозную кровь.
Как правильно подготовиться к исследованию?
Подготовки не требуется.
Подробнее об исследовании
До 10 % людей имеют предрасположенность к инфекционным заболеваниям из-за недостаточности важной составляющей системы врождённого иммунитета – манноз-связывающего лектина (МСЛ).
Манноз-связывающий лектин синтезируется в печени и участвует в иммунной защите. Этот белок связывается с сахарами на поверхности микроорганизмов, таким образом запускается один из основных путей активации системы комплемента – лектиновый путь, что способствует удалению патогенов с помощью комплемент-опосредованного фагоцитоза.
В реакции принимают участие белки, которые работают вместе для уничтожения бактерий и других антигенов. МСЛ циркулирует в крови в неактивной форме. Когда он активирован, он приводит в движение цепную реакцию, конечные продукты которой способны уничтожать бактерии и другие антигены (вирусы, грибы).
Эпидемиологические исследования показали, что от концентрации МСЛ в сыворотке крови зависит чувствительность к различным инфекциям, а также предрасположенность к аутоиммунным (в частности, ревматоидному артриту), метаболическим и сердечно-сосудистым заболеваниям.
При снижении концентрации и изменении структуры манноз-связывающего белка в сыворотке увеличивается риск развития тяжелых форм бактериальных и вирусных инфекций у детей и взрослых (пневмонии, отита, сепсиса, хронической диареи), повышается восприимчивость к гепатитам В и С, ВИЧ, характерно затяжное течение инфекционно-воспалительных заболеваний с рецидивами. Большее влияние дефицит лектина оказывает на сопротивляемость инфекциям у детей в возрасте 6-18 месяцев. Количество МСЛ ассоциировано с развитием инфекционных осложнений у ВИЧ-инфицированных и онкологических больных, получающих химиотерапевтические препараты. Для больных муковисцидозом течение болезни и прогноз также может зависеть от содержания МСЛ.
Уровень МСЛ на 96 % определяется генетическими факторами. Его дефицит может возникнуть в результате одного или нескольких изменений в гене MBL2. В данное исследование включено шесть генетических маркеров, влияющих на уровень МСЛ в сыворотке крови. Генетические маркеры расположены в 1-м экзоне, который кодирует N-терминальный домен белка, и в промоторной области гена MBL2. В 1-м экзоне гена выявлено три точки, в которых могут наблюдаться нуклеотидные замены: 1) в позиции +154 замена азотистого основания цитозина на тимин C/T (Arg52Cys), 2) в позиции +161 замена гуанина на аденин G/A (Gly54Asp), 3) в позиции +170 замена гуанина на аденин G/A (Gly57Glu). Они обозначаются как D, B и C соответственно, а измененный по любому из этих маркеров аллель – как О. Генотип A/A (wildtype, неизмененные аллели) характерен для людей с нормальной концентрацией МСЛ в сыворотке. Замены также могут располагаться в промоторной области гена в позициях –550G/C (H/L вариант), –221G/C (Y/X вариант), и в 5'-нетранслируемом регионе +4C/T (P/Q вариант). Различают семь гаплотипов: HYPA, LYQA, LYPA, LXPA, LYPB, LYQC и HYPD.
Изменения в структуре MBL2 нарушают маннозный путь активации комплемента. Специфических методов лечения таких нарушений в настоящее время не существует. В критических ситуациях для замены компонентов комплемента может быть использована свежезамороженная плазма. В отдельных случаях можно рекомендовать вакцинацию (например, менингококковую, пневмококковую вакцины).
Определение гаплотипа MBL2 позволит своевременно начать профилактику инфекционных заболеваний, а также, что не менее важно, при наличии клинических симптомов определиться с их причинами.
В будущем эффективным методом лечения может быть генная терапия (заместительная терапия рекомбинантным белком МСЛ).
Когда назначается исследование?
- При тяжелых, рецидивирующих инфекциях у детей и взрослых.
- При частых инфекционных заболеваниях с тяжелым течением у детей, после периода грудного вскармливания.
- При повторных инфекциях у детей раннего и младшего возраста.
- При выявлении повышенной восприимчивости к гепатиту В и С, ВИЧ-инфекции.
- При составлении прогноза инфекционных осложнений у ВИЧ-инфицированных пациентов.
- При составлении прогноза инфекционных осложнений у пациентов с онкопатологией на фоне применения химиотерапии.
- При составлении прогноза заболевания и при оценке риска осложнений у больных муковисцидозом.
- При составлении прогноза инфекционных осложнений у пациентов с бронхиальной астмой, хронической обструктивной болезнью легких.
Что такое генетические болезни? Обременительное наследство
Для начала необходимо разобраться в терминах. Начнем с того, что генетические заболевания и заболевания, к которым выявлена наследственная предрасположенность, — разные понятия.
Теперь разберемся в механизме наследования. Формируясь, зародыш получает половину хромосом от матери, а половину — от отца. Именно поэтому организм ребенка не копирует ни одного из родителей, а имеет свою индивидуальность. Передача хромосом, генов, а значит, и передача информации о наследственных заболеваниях, возможна по нескольким схемам:
К наиболее распространенным генетическим заболеваниям относятся:
- дальтонизм — около 850 случаев на 10 000;
- расщепление позвоночника — 10–20 случаев на 10 000 человек;
- синдром Клайнфельтера (эндокринные нарушения, которые могут стать причиной мужского бесплодия) — 14–20 на 10 000;
- синдром Дауна — 9–13 на 10 000;
- синдром Тернера (болезнь, которая приводит к половому инфантилизму) — около 7 на 10 000;
- фенилкетонурия (нарушение метаболизма аминокислот) — до 3,8 на 10 000;
- нейрофиброматоз (заболевание, при котором у больного возникают опухоли) — около 3 на 10 000;
- муковисцидоз — 1–5 на 10 000;
- гемофилия — до 1,5 на 10 000 [8] .
Сегодня врачи выявляют генетические заболевания с высокой точностью, так как передовые технологии позволяют буквально заглянуть внутрь гена, определить, на каком уровне произошло нарушение.
Есть несколько направлений обследований.
Диагностическое тестирование проводится, если у пациента есть симптомы или особенности внешнего развития, служащие отличительной чертой генетического заболевания. Перед направлением на диагностическое тестирование проводят всесторонний осмотр пациента. Одна из отличительных черт наследственных заболеваний — это поражение нескольких органов и систем [10] , поэтому при выделении целого ряда отклонений от нормы врач направляет пациента на молекулярно-генетическую диагностику.
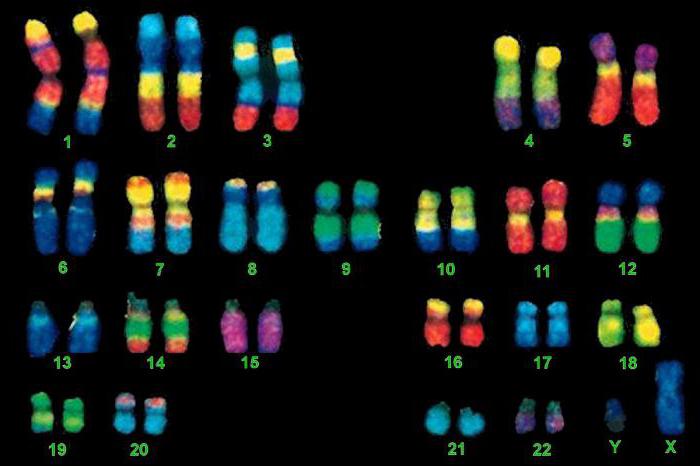
Так как многие наследственные заболевания (например, синдромы Дауна, Эдвардса, Патау) связаны с нарушением количества хромосом (кариотипа), то для их подтверждения проводят кариотипирование, то есть изучение количества хромосом. Для анализа требуются клетки крови, которые в течение нескольких дней выращивают в особой среде, а затем окрашивают. Так врачи выделяют и идентифицируют каждую хромосому, определяют, нарушен ли их количественный состав [11] , отмечают особенности внешнего строения.
Для выявления мутаций конкретных генов применяется метод ПЦР — полимеразной цепной реакции. Его суть состоит в выделении ДНК и многократном воспроизводстве интересующего исследователя участка. Как отмечают специалисты, преимущество ПЦР — его высокая точность: здесь почти невозможно получить ложноположительный результат. Метод удобен еще и тем, что для исследования может быть взята любая ткань организма [12] .
Если вы знаете, что у вас в семье или в семье супруга были случаи наследственных болезней, то, конечно, захотите выяснить, какова вероятность проявления их у ваших детей. Врачи часто предлагают будущим родителям сделать пренатальную диагностику. А если пара использует вспомогательные репродуктивные технологии, то и предимплантационную генетическую диагностику плода (ПГД).
Пренатальная генетическая диагностика проводится, когда ребенок еще находится в утробе матери. Предположить наличие генетических отклонений врач может на основании анализов крови матери или по результатам УЗИ плода. Поэтому на начальном этапе беременная проходит трехмаркерный скрининг: в ее крови определяют уровень АФП, β-хорионического гонадотропина и эстриола. Если их концентрация отлична от нормы, то врач рекомендует выполнить генетическое обследование ребенка. Для этого с помощью пункции берут амниотическую жидкость и проводят кариотипирование плода. Единственный недостаток этого метода — долгий период ожидания результатов. Если последний будет негативным, то женщина просто может не успеть принять решение о прерывании беременности. Есть и альтернатива — анализ ворсин хориона. Его можно сделать на раннем сроке, но получение материала представляет угрозу для протекания беременности [14] .
В последнее время появилась еще одна возможность пренатального обследования плода — неинвазивный пренатальный ДНК-тест (НИПТ-тест). В этом случае нужна только кровь матери. Точность теста достигает 99%, причем можно сделать обследование как на самые часто встречающиеся генетические патологии, так и полное исследование плода [15] .
Рассматривая виды наследования генетических заболеваний, мы упомянули об аутономно-рецессивном способе и о наследовании, сцепленном с полом. Человек может быть здоров, но в его генотипе при этом присутствует патологический ген. Выявить это помогает анализ на носительство. Многие делают его на стадии планирования беременности, чтобы вычислить вероятность рождения ребенка с генетическими заболеваниями.
Например, такая болезнь, как гемофилия, проявляется только у мужчин, женщины не болеют, но могут быть носителями. Поэтому женщинам, у которых есть родственники с проблемами свертывания крови, перед зачатием рекомендуется сделать скрининг гетерозиготного носительства, чтобы определить вероятность рождения мальчика с гемофилией [16] .
По итогам ДНК-идентификации врач дает пациенту рекомендации: начиная от образа жизни и диеты и заканчивая профессиональными рисками. Следование им помогает избежать развития многих заболеваний.
В зависимости от того, чем вызвано генетическое заболевание, врач выбирает и методы обследования пациента. Рассмотрим основные группы патологий.
Причиной этих генетических заболеваний служит нарушение в количественном составе хромосом или в их строении. Например, при наличии дополнительной (третьей) 21-й хромосомы формируется синдром Дауна. Причиной синдрома Шершевского-Тернера является наличие всего одной Х-хромосомы у женщин. А если у мужчины половые хромосомы присутствуют в сочетании XXY, а не XY, то ему ставится синдром Клайнфельтера.
Для диагностики проводят кариотипирование. В качестве примера можно привести синдром Клайнфельтера — редкое генетическое заболевание, которым страдают мужчины. Внешне оно выражается в евнухоподобной внешности, увеличении грудных желез, нарушении половой функции. Подробное изучение состава половых хромосом помогает определить, какое именно нарушение произошло у пациента (лишних Х-хромосом может быть несколько). В зависимости от кариотипа варьируется и степень выраженности признаков заболевания [20] .
Пациентам с хромосомными заболеваниями назначают цитогенетическое обследование. Обычно ему подвергаются и родители, чтобы установить, имеет ли место наследуемая патология или же это единичный случай [22] .
Нарушения могут произойти не в хромосоме, а лишь на одном ее участке. Тогда мы говорим о генной мутации. Эти заболевания называются моногенными, к ним, в частности, относятся многие нарушения метаболизма: муковисцидоз, фенилкетонурия, андрогенитальный синдром и т.д. Многие из этих заболеваний могут быть выявлены при обязательном скрининге всех младенцев в роддоме. Ребенок, у которого есть отклонения от нормы, может быть направлен на дополнительное генетическое обследование. А принятые вовремя меры позволяют в некоторых случаях предотвратить развитие серьезных нарушений.
В то же время существуют заболевания, вызванные генными мутациями, которые не проявляются ярко и однозначно. В качестве примера можно привести синдром Вольфрама, который дебютирует как сахарный диабет в раннем возрасте, затем проявляется ухудшением зрения или слуха. Врач может подтвердить синдром только по результатам генетической экспертизы.
Они выявляются при ДНК-идентификации. Анализ подтверждает наличие или отсутствие предрасположенности практически к любой патологии: от сахарного диабета до формирования различных зависимостей [23] . Так как роль генетических факторов и факторов внешней среды в развитии заболеваний различна не только для каждой патологии, но и для каждого пациента [24] , рекомендации здесь могут быть только строго индивидуальными, сделанными на основании результатов анализов.
Наследственные заболевания отличаются большим разнообразием: это могут быть патологии, вызванные мутацией генов, нарушением строения хромосом, сочетанием нескольких факторов, в том числе факторов внешней среды. Именно поэтому генетическое обследование лучше выполнять в лаборатории, которая предоставляет максимально широкий спектр услуг. Желательно, чтобы в лаборатории проводилось и кариотипирование, и ПЦР, и пренатальная диагностика, и анализ на носительство.
Второй важный момент — наличие в лаборатории современного сертифицированного оборудования. Оно позволяет делать анализ максимально подробным и полным. Популярные экспресс-системы дают результат в тот же день, однако глубокий анализ генотипа им недоступен. Специализированные лаборатории предоставляют результаты через 2–3 дня, однако это более подробное и детализированное исследование, позволяющее точно установить и наличие заболевания, и предрасположенность к тем или иным патологиям.
Стоимость обследования в специализированной лаборатории во многом зависит от объема: при составлении генетического паспорта цена обследования может достигать 75 000–80 000 рублей [26] .
[youtube.player]Полный текст:
В обзоре рассматриваются вопросы, связанные с генетической предрасположенностью и устойчивостью к инфекционным заболеваниям. Генетические факторы в значительной мере определяют восприимчивость организма к различным заболеваниям, в том числе к инфекционным. Показана генетическая предрасположенность к туберкулезу, сальмонеллезу, вирусным гепатитам, клещевому энцефалиту, болезни Лайма, ВИЧ и другим. Знание молекулярно-генетических биомаркеров необходимо для выделения групп риска, проведения предиктивных мероприятий, в частности вакцинации. Основное влияние уделяется генам главного комплекса гистосовместимости, показана роль митохондриальной ДНК в восприимчивости к ВИЧ-инфекции.
д. б. н., проф., зав. кафедрой клинической лабораторной диагностики ФДПО
к. б. н., доцент кафедры, кафедра клинической лабораторной диагностики ФДПО
д. м. н., проф., зав. кафедрой кожных и венерических болезней и косметологии ФНМО Медицинского института
д. б. н., проф., проф. кафедры, кафедра клинической лабораторной диагностики ФДПО
д. б. н., проф., проф. кафедры, кафедра клинической лабораторной диагностики ФДПО
1. Morens D. M., Folkers G. K., Fauci A. S. The challenge of emerging and re-emerging infectious diseases. // Nature. — 2004. — 430. — 6996. — P. 242–249.
2. DeWitte S. N. Mortality risk and survival in the aftermath of the medieval Black Death. // PLoSOne. — 2014. — 9. — 5: e96513.
3. Kumar V., Wijmenga C., Xavier R. J. Genetics of immune-mediated disorders: from genome-wide association to molecular mechanism. // Curr. Opin. Immunol. — 2014. — 31. — P. 51–57.
4. Boisson-Dupuis S., Bustamante J., El-Baghdadi J. et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. // Immunol. Rev. — 2015. — 264. — P. 103–120.
5. Amos W., Driscoll E., Hoffman J. I. Candidate genes versus genome-wide associations: which are better for detecting genetic susceptibility to infectious disease? // Proc. Biol. Sci. — 2011. — 278. — P. 1031–1037.
6. Burton P. R., Hansell A. L., Fortier I. et al. Size matters: just how big is BIG?: Quantifying realistic sample size requirements for human genome epidemiology. // Int. J. Epidemiol. — 2009. — 38. — P. 263–273.
7. Newport M. J., Finan C. Genome-wide association studies and susceptibility to infectious diseases. // Brief Funct Genomics. — 2011. — 10. — P. 98–107.
8. Loeb M., Eskandarian S., Ropp M. et al. Genetic variants and susceptibility to neurological complications following West Nile virus infection. // J. Infect. Dis. — 2011. — 204. — P. 1031–1037.
9. Matzaraki V., Kumar V., Wijmenga C. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. // Genome Biol. — 2017. — 18: 76.
10. Cortes A, Brown MA. Promise and pitfalls of the Immunochip. // Arthritis Res Ther. 2011; 13: 101. DOI: 10.1186/ar3204.
11. Carapito R, Radosavljevic M, Bahram S. Next-generation sequencing of the HLA locus: methods and impacts on HLA typing, population genetics and disease association studies. // Hum. Immunol. — 2016. — 77. — 11. — P. 1016–1023.
12. Nagasaki M., Yasuda J., Katsuoka F. et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. // Nat Commun. — 2015; 6: 8018. DOI: 10.1038/ncomms9018.
13. Jain M., Koren S., Miga K. H. et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. // Nat. Biotechnol. — 2018. — 36. — 4. — P. 338–345.
14. Хаитов Р. М., Алексеев Л. П., Трофимов Д. Ю. Иммуногеномика и генодиагностика человека. Национальное руководство. ГЭОТАР-Медиа. — М. — 2017–256 с.
15. Fellay J., Shianna K. V., Ge D. et al. A Whole-Genome Association Study of Major Determinants for Host Control of HIV-1. // Science. — 2007. — 10.1126/science.1143767.
16. Fellay J., Shianna K. V., Ge D. et al. A whole-genome association study of major determinants for host control of HIV-1. // Science. — 2007. — 317. P. 944–947.
17. Fellay J., Ge D., Shianna K. V., Colombo S. et al. Common genetic variation and the control of HIV-1 in humans. // PLoS Genet. — 2009. — 5. DOI: 10.1371/journal.pgen.1000791.
18. Limou S., Le Clerc S., Coulonges C.et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) // J. Infect. Dis. — 2009. —199. — P. 419–26.
19. International HIV Controllers Study. Pereyra F., Jia X., McLaren P.J. et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. // Science. — 2011. —330. — P. 1551–1557.
20. McLaren P.J., Coulonges C., Ripke S. et al. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. // PLoS Pathog. — 2013. — 9. — 7. doi: 10.1371/journal.ppat.1003515.
21. Hendrickson S. L., Hendrickson S. L., Ruiz-Pesini E. et al. Mitochondrial DNA haplogroups influence AIDS progression // AIDS. — 2008. — 22. — 18. — P. 2429–2439.
22. Chinnery P. F., Elliott H. R., Syed A. Mitochondrial DNA haplogroups and risk of transient ischaemic attack and ischaemic stroke: a genetic association study. // Lancet Neurol. — 2010. — 9. — 5. — C. 498–503.
[youtube.player]От родителей ребенок может приобрести не только определенный цвет глаз, рост или форму лица, но и генетические заболевания, передающиеся по наследству. Какие они бывают? Как можно их обнаружить? Какая классификация наследственных болезней существует?
Механизмы наследственности
Прежде, чем говорить о заболеваниях, стоит разобраться, что такое генетическая наследственность. Вся информация о нас содержится в молекуле ДНК, которая состоит из невообразимо длинной цепочки аминокислот. Чередование этих аминокислот уникально.
Фрагменты цепочки ДНК называются генами. В каждом гене заключается целостная информация об одном или нескольких признаках организма, которая передается от родителей детям, например, цвет кожи, волос, черта характера и т. д. При их повреждении или нарушении их работы возникают генетические заболевания, передающиеся по наследству.
ДНК организовано в 46 хромосомах или 23 парах, одна из которых является половой. Хромосомы отвечают за активность генов, их копирование, а также восстановление при повреждениях. В результате оплодотворения в каждой паре присутствует одна хромосома от отца, а другая от матери.
При этом один из генов будет доминантным, а другой рецессивным или подавляемым. Упрощенно, если у отца ген, отвечающий за цвет глаз, окажется доминантным, то ребенок унаследует этот признак именно от него, а не от матери.
Генетические заболевания
Передающиеся по наследству болезни возникают, когда в механизме хранения и передачи генетической информации происходят нарушения или же мутации. Организм, чей ген поврежден, будет передавать его своим потомкам точно так же, как и здоровый материал.
В том случае, когда патологический ген является рецессивным, он может и не проявляться у следующих поколений, но они будут его переносчиками. Шанс, что доминантный ген не проявится, существует, когда здоровый ген тоже окажется доминантным.
В настоящее время известно больше 6 тысяч наследственных заболеваний. Многие из них проявляются после 35 лет, а некоторые могут никогда не заявить о себе хозяину. С крайне высокой частотой проявляется сахарный диабет, ожирение, псориаз, болезнь Альцгеймера, шизофрения и другие расстройства.
Классификация
Генетические заболевания, передающиеся по наследству, имеют огромное количество разновидностей. Для разделения их на отдельные группы может учитываться локация нарушения, причины, клиническая картина, характер наследственности.
Болезни могут классифицироваться по типу наследования и локации дефектного гена. Так, важно, расположен ген в половой или неполовой хромосоме (аутосоме), а также является он подавляющим или нет. Выделяют заболевания:
- Аутосомно-доминантные – брахидактилия, арахнодактилия, эктопия хрусталика.
- Аутосомно-рецессивные – альбинизм, мышечная дистония, дистрофия.
- Ограниченные полом (наблюдаются только у женщин или мужчин) – гемофилия А и Б, цветовая слепота, паралич, фосфат-диабет.
Количественно-качественная классификация наследственных болезней выделяет генные, хромосомные и митохондриальные виды. Последний относится к нарушениям ДНК в митохондриях за пределами ядра. Первые два происходят в ДНК, которая находится в ядре клетки, и имеют несколько подвидов:
Мутации или отсутствие гена в ядерной ДНК.
Синдром Марфана, адреногенитальный синдром у новорожденных, нейрофиброматоз, гемофилия А, миопатия Дюшенна.
Предрасположенность и действие экзогенных факторов.
Псориаз, шизофрения, ишемическая болезнь, цирроз, бронхиальная астма, сахарный диабет.
Изменение структуры хромосом.
Синдромы Миллера-Диккера, Вильямса, Лангера-Гидиона.
Изменение числа хромосом.
Синдромы Дауна, Патау, Эдвардса, Клайфентера.
Причины возникновения
Наши гены склонны не только накапливать информацию, но и изменять её, приобретая новые качества. Это и есть мутация. Происходит она довольно редко, примерно 1 раз на миллион случаев, и передается потомкам, если произошла в половых клетках. Для отдельных генов частота мутации составляет 1:108.
Мутации являются естественным процессом и составляют основу эволюционной изменчивости всех живых существ. Они могут быть полезными и вредными. Одни помогают нам лучше приспособиться к окружающей среде и способу жизни (например, противопоставленный большой палец руки), другие приводят к заболеваниям.

Возникновение патологий в генах учащают физические, химические и биологические мутагенные факторы. Таким свойством обладают некоторые алкалоиды, нитраты, нитриты, некоторые пищевые добавки, пестициды, растворители и нефтяные продукты.
Среди физических факторов находятся ионизирующие и радиоактивные излучения, ультрафиолетовые лучи, чрезмерно высокие и низкие температуры. В качестве биологических причин выступают вирусы краснухи, кори, антигены и т. д.
Генетическая предрасположенность
Родители влияют на нас не только воспитанием. Известно, что одни люди имеют больше шансов появления некоторых заболеваний, чем другие из-за наследственности. Генетическая предрасположенность к заболеваниям возникает, когда кто-то из родственников имеет нарушения в генах.
Риск возникновения конкретного заболевания у ребенка зависит от его пола, ведь некоторые болезни передаются только по одной линии. Он также зависит от расы человека и от степени родства с больным.
Если у человека с мутацией рождается ребенок, то шанс унаследования болезни будет 50%. Ген вполне может никак себя не проявить, будучи рецессивным, а в случае брака со здоровым человеком, его шансы передаться потомкам составят уже 25%. Однако если супруг тоже будет владеть таким рецессивным геном, шансы проявления его у потомков снова увеличатся до 50 %.
Как выявить болезнь?
Вовремя обнаружить заболевание или предрасположенность к нему поможет генетический центр. Обычно такой есть во всех крупных городах. Перед сдачей анализов проводится консультация с врачом, чтобы выяснить, какие проблемы со здоровьем наблюдаются у родственников.
Медико-генетическое обследование проводится путем взятия крови на анализ. Образец внимательно изучается в лаборатории на предмет каких-либо отклонений. Будущие родители обычно посещают подобные консультации уже после наступления беременности. Однако в генетический центр стоит прийти и во время её планирования.

Наследственные заболевания серьезно отражаются на психическом и физическом здоровье ребенка, влияют на продолжительность жизни. Большинство из них тяжело поддается лечению, а их проявление только корректируется медицинскими средствами. Поэтому лучше подготовиться к подобному ещё до зачатия малыша.
Синдром Дауна
Одна из наиболее распространенных генетических болезней – синдром Дауна. Она встречается в 13 случаях из 10000. Это аномалия, при которой человек имеет не 46, а 47 хромосом. Диагностировать синдром можно сразу при рождении.
Среди главных симптомов уплощенное лицо, приподнятые уголки глаз, короткая шея и недостаток мышечного тонуса. Ушные раковины, как правило, маленькие, разрез глаз косой, неправильная форма черепа.

У больных детей наблюдаются сопутствующие расстройства и болезни – пневмония, ОРВИ и т. д. Возможно возникновение обострений, например, потеря слуха, зрения, гипотериоз, заболевания сердца. При даунизме умственное развитие замедлено и часто остается на уровне семи лет.
Постоянная работа, специальные упражнения и препараты значительно улучшают ситуации. Известно много случаев, когда люди с подобным синдромом вполне могли вести самостоятельную жизнь, находили работу и достигали профессиональных успехов.
Гемофилия
Редкое наследственное заболевание, поражающее мужчин. Встречается один раз на 10 000 случаев. Гемофилия не лечится и возникает в результате изменения одного гена в половой Х-хромосоме. Женщины являются только переносчиками болезни.
Основной характеристикой является отсутствие белка, который отвечает за свертывание крови. В таком случае, даже незначительная травма вызывает кровотечение, которое не просто остановить. Иногда оно проявляет себя только на следующий день после ушиба.
Синдром Ангельмана

Синдром возникает раз на 10 000 случаев из-за отсутствия некоторых генов в длинном плече 15-й хромосомы. Болезнь Ангельмана развивается только, если гены отсутствуют в хромосоме, доставшейся от матери. Когда те же гены отсутствуют в отцовской хромосоме, возникает синдром Прадера-Вилли.
Заболевание нельзя излечить полностью, но облегчить проявление симптомов возможно. Для этого проводятся физические процедуры и массажи. Полностью самостоятельными больные не становятся, но при лечении могут сами себя обслуживать.
[youtube.player]Болезни с наследственным предрасположением отличаются от описанных выше энзимопатий тем, что их проявление, хотя и обусловлено генетическими факторами, в значительной степени зависит от провоцирующих их проявление факторов внешней среды. Исходя,из первично генетической природы их обычно делят на 2 группы - моногенные и полигенные (мультифакториальные) болезни.
Первые из них характеризуются тем, что в основе болезни лежит мутация того или иного гена, но для их проявления требуется действие конкретного внешнего агента, который обычно можно точно установить. По отношению к данной болезни он может рассматриваться как специфический.
Полигенные болезни с наследственным предрасположением обусловлены значительно более сложным взаимодействием многих генов - полигенов. При этом каждый из них является скорее нормальным, чем патологическим и свое действие полигены
осуществляю'! с комплексом факторов внешней среды. При этом относительная роль генетических и средовых факторов различна не только для данной болезни, но для каждого индивидуального случая заболевания,
3.2.1. Моногенные болезни с наследственным предрасположением относительно немногочисленны, К ним вполне применимы методы менделевского генетического анализа, а их профилактика и лечение достаточно разработаны и обычно эффективны. Иначе говоря, в таких случаях речь идет как бы о замаскированных генах, которые проявляют себя только в определенных условиях. Примерами таких болезней могут служить случаи развития каких-либо патологических состояний у лиц, вынужденных принимать те или иные лекарства, необычные реакции на те или иные продукты питания, факторы внешней среды химического и физического характера и т.д.. Эти обстоятельства обуславливали появление таких направлений в развитии медицинской генетики, как фармагенетика и экогенетика.
Классический пример-так называемая примахиновая анемия
или фавизм. В основе патогенеза этого заболевания лежит гемолиз
эритроцитов, наступающий лишь у некоторых людей после приема
ряда лекарственных препаратов (всего около 30 наименований, в
том числе и противомалярийного препарата примахина,
сульфаниламидов) или употребления в пищу конских бобов Viola
lava. Генетическая основа- мутация гена, контролирующего синтез
фермента глкжозо-6-фосфатдегидрогеназы (Г-6-ФДГ).
11оследствием этой мутации является снижение активности данного фермента, особенно в стареющих эритроцитах, что и обуславливает их распад - гемолиз. Заболевание наследуется сцепленно с X-хромосомой по рецессивному типу. Обладатели данной мутации в гомо- и гетерозиготном состоянии устойчивы к малярии, в связи с чем она достаточно широко распространена в ряде популяций Африки, Азии, Средиземноморья, Закавказья.
Приведем еще несколько примеров. Катехоламины, содержащиеся в сыре, могут вызвать мигрень у некоторых индивидов. Другим пищевым продуктом, провоцирующим мигрень, является шоколад, что объясняется низкой активностью фермента моноаминооксидазы у таких лиц.
Известны специфические реакции людей на алкоголь. В популяциях азиатских народов они встречаются особенно часто. У лиц, принимающих небольшие /юзы алкоголя, наблюдается
покраснение лица и развиваются признаки алкогольного отравления, что связано с наследственными вариациями в ферментах (алкогольдегидрогеназы), расщепляющих спирт.
Ряд лиц не переносят жирной пищи и раннем возрасте страдают атеросклерозом, инфарктом миокарда. Гены непереносимости лактозы широко распространены в популяциях Юго-Восточной Азии (до 95 - 100% корейцев, китайцев, японцев), среди американских негров и индейцев (70 - 75%). Суть этого дефекта сводится к отсутствию выработки в кишечнике фермента лактазы, при этом ферментативное расщепление лактазы заменяется бактериальным, что ведет к развитию поносов. Некоторые дети страдают синдромом нарушенного всасывания в кишечнике в связи с непереносимостью белка клейковины пшеницы и ржи - глиадина. Заболевание носит название цилиакии. При этом нарушатся всасывание в тонком кишечнике воды, жиров, углеводов, белков, что сопровождается поносами, потерей веса, повышенным выведением азота и другими проявлениями. Наследуется по аутосомно-доминантному типу с неполной ' пенетрантностью.
Примерами различных реакций на физические факторы внешней среды может являться индивидуальная чувствительность к теплу, холоду, солнечному свету, солям тяжелых металлов и т.д..
Как видно из выше изложенного, эту группу болезней также относят к энзимопатиям, но клиническое проявления наступают лишь в случаях действия провоцирующих факторов, устранение которых оказывает терапевтический эффект.
3.2.2. Мультифакториальные болезни составляют 90% хронических неинфекционных болезней различных систем и органов человека. В этиологии и патогенезе данной категории болезней принимают участие, как многие гены, так и разнообразные средовые факторы, в связи с чем генетический анализ по конкретизации роли индивидуальных генов и их взаимодействие со средовыми факторами оказывается весьма трудной задачей, которую еще предстоит решать (Табл. №1 1>.
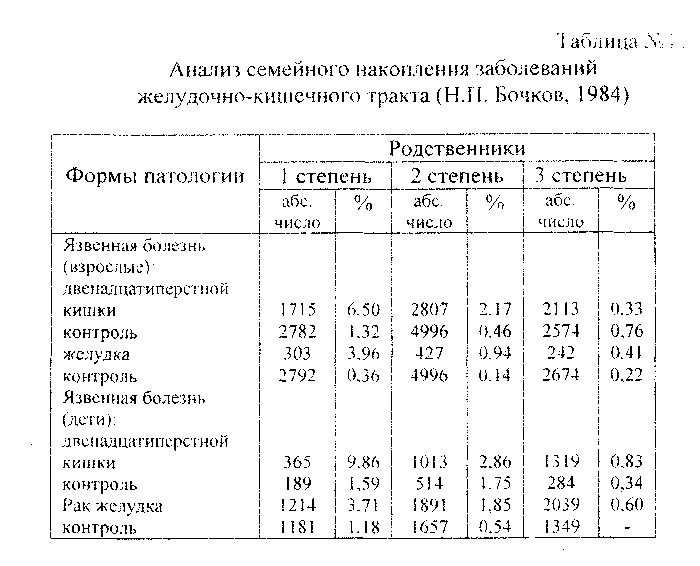
клиническими симптомами (например, диабет, язвенная болезнь желудка, шизофрения и многие другие психические заболевания, по-видимому, большинство кожных болезней, атеросклероз и др.). Наиболее часто используемые методы изучения этой группы болезней — клинико-генеалогичеекий, близнецовый, популяционно-статистический, биохимический, иммунологический с широким привлечением компьютерных и математических методов анализа полученных результатов.
Заключая данный раздел, можно сказать, что все мультифакториальные болезни могут рассматриваться как примеры экогенетики человека, поскольку их развитие является результатом взаимодействия генов предрасположенности и факторов внешней среды.
Хромосомные болезни
К хромосомным болезням относятся врожденные формы патологии, которые клинически обычно выражаются множественными пороками развития, а в качестве генетической основы имеют отклонения от нормального содержания в клетках количество хромосомного материала, т.е. обусловлены геномными мутациями или хромосомными аберрациями.
Большинство хромосомных болезней не передаются по наследству, а возникают заново вследствие мутаций в предшественниках гамет здоровых родителей или первых делениях зиготы. Такие зиготы с несбалансированным набором хромосом обычно погибают, что влечет за собой спонтанный выкидыш. В среднем около 40% диагностируемых спонтанных выкидышей обусловлены ' хромосомным дисбалансом, около 6% мертворожденных детей имеют хромосомные изменения. На 1000 живорожденных - 3 - 4 имеют хромосомные болезни. Если все случаи множественных пороков развития принять за 100%, то 35 -40% из них будут приходится на хромосомные болезни.
По характеру изменений в генотипе все хромосомные болезни можно четко разделить на две группы.
Первая группа - геномные четные, при которых происходит изменение плоидности хромосом кратно геномному числу (гаплоидия и полиплоидия) и геномные нечетные, когда изменяется количество хромосом в отдельных парах на одну или несколько (анеуплоидии. гетероплоидии).
Вторая группа - структурно-хромосомные, обусловленные хромосомными аберрациями всех типов (инверсии, дупликации. транслокации, делении).
У человека описаны тришюидные и тетраплоидные наборы хромосом у абортированных плодов, эмбрионов и мертворожденных. Продолжительность жизни новорожденных с такой патологией - несколько дней. Среди живущих описаны лишь мозаичные формы, при которых часть соматических клеток полиплоидна, а часть -диплоидна.
Гетероплоидии по отдельным парам хромосом достаточно многочисленны (Табл. №121.


Полные моносомии (полностью отсутствует одна из хромосом) среди живых наблюдается только по X - хромосоме (45, X). Однако в среднем 29 из 30 зигот с таким набором погибает. Полная моносомия по любой из пар аутосом ведет к эмбриолетальности. Среди живых описаны лишь мозаики.
Полные гриеомии описаны для многих пар хромосом: 8. 9. 13, 14, 21, 22. X - хромосомам. Число X - хромосом может доходить у отдельных индивидов с сохранением их жизнеспособности до 5 .
Изменения числа хромосом вызываются нарушением их распределения по дочерним клеткам во время 1-го (редукционного) или 2-го (эквационного) мейотического деления или в первых дроблениях зиготы (Рис. 32). Если такие нарушения происходят в двух и более последовательных делениях, то возникают тетра-, лента- и другие полисомии.
Структурные перестройки хромосом (аберации), какого бы вида они не были (недостатки - делении или избытка- дупликации генетического материала), вызывают нарушения развития. Такого рода изменения генотипа носят название частичных моно- или трисомий. Небезобидны и инверсии, при которых гены меняют свое положение в хромосоме на 180°.
Существенную роль в развитии генетической патологии у человека играют транслокации. Они бывают сбалансированными, не приносящими вреда обладателю такой мутации, но чреваты генетическими последствиями для потомства, формируя несбалансированную зиготу, которая или гибнет на том или ином этапе эмбриогенеза (что бывает чаще всего), или ее развитие заканчивается рождением больного ребенка.
Ниже мы приводим краткое клинико-генетическое описание некоторых из наиболее часто встречающихся хромосомных синдромов человека.
3.3.1. Болезнь Дауна
Впервые болезнь Дауна была описана английским педиатром Л. Дауном в 1866 г., как синдром монголоидной идиотии. Лишь в 1959 г. установлено, что у детей, страдающих данной патологией, имеет место трисомия по 21 хромосоме (генотип - 47 XX, 21+). Внешний вид больных обычно настолько типичен, что можно говорить о портретном узнавании больных, а не о диагностическом понимании. У большинства больных плоское лицо, монголоидный разрез глаз, эпикант, открытый рот (относительно увеличенный язык Tie вмещается в ротовой полости), короткий нос с плоской переносицей, нередко косоглазие, брахицефалия, аркообразное небо, разнообразные зубные аномалии. Типичны множественные пороки развития желудочно-кишечного тракта и сердца. В ряде
случаев болезнь Дауна ассоциируется с эпилепсией и лейкозами. Интеллект всегда в той или иной мере значительно снижен. Продолжительность жизни зависит от тяжести пороков развития внутренних органов, в основном сердечно-сосудистой системы. При современном уровне развития медицины многие из них доживают до 20 - 40 - летнего возраста.

Репродуктивные функции у больных сохраняются. В литературе описано более 20 случаев рождения женщинами с болезнью Дауна детей. Гак. из 13 учтенных беременностей у 11 женщин с трисомией по 21 хромосоме, 5 закончились рождением детей с болезнью Дауна и 7 - рождением нормальных (в одном случае имела место двуплодная беременность). Риск повторного рождения ребенка с болезнью Дауна одной и той женщиной (при простой трисомии) зависит от возраста мамы. Он колеблется от 1: 800 до 30 лет и до I: 25 после 44. Возраст отца также влияет на частоту рождения больных детей.
Различают так называемую регулярную форму синдрома, которая вызывается простой трисомией случаев болезнью Дауна, и транслокационную (Рис. 33, 34). Наиболее распространена простая трисомная форма синдрома Дауна (94%). транслокационная форма составляет 4% (принцип транслокации хромосом показан на рис.35), мозаичная - 2%. Популяционная, частота - 1 ' :700. Фенотипический эффект при обеих формах одинаковый. Дри первой из них мутации возникают каждый раз заново (de novo).

Рис. 35. Принцип центрического слияния (Робертсоновская транслокация). Две акроцентрические хромосомы утратили свои короткие плечи (р), а длинные плечи (q) слились. Индивид, имеющий такую транслокацию, будет иметь на одну хромосому меньше по сравнению с нормой, однако это не приводит к каким-либо функциональным отклонениям и носители таких хромосом совершенно здоровы.

т.е. у родителей больного ребенка нормальный набор хромосом. При второй - родители здоровы фенотипически, но у кого-то из них имеет место сбалансированная транслокация 21 хромосомы, а чаще ее длинных плеч, на любую другую хромосому той же группы (№21 и №22) или с любой другой. В случае транслокации 21 на 21 во всех случаях будут формироваться только патологические гаметы. Варианты образования гамет при транслокации №21 на хромосому №15 показано на Рис.36. После оплодотворения нормальным сперматозоидом (15, 21) получим результаты, которые хорошо демонстрирует решетка Пеннета (Рис.37).
При мозаичном варианте выраженность клинических симптомов зависит от соотношения нормального и патологического клонов клеток: чем меньше процент нормальных клеток с 46 хромосомами, тем более выражена клиническая симптоматика. Напротив, при высоком содержании нормальных клеток клинические проявления стерты, и в этом случае необходимо цитогенетическое обследование.
У больных с синдромом Дауна изменен угол трирадиуса atd (Рис.38).

3,3.2. Синдром трисомии13
Впервые трисомия 13 хромосомы (47, XX, 13+) была описана К. Патау в 1960 г. и названа в его честь синдромом Патау. Это была девочка, родившаяся у 25-летней женщины, перенесшей на шестом месяце беременности грипп. Ребенок имел масс> пороков развития микроцефалию, анафтальмию (отсутствовали глаза). двустороннюю расщелину губы, полное незаращение неба, иолидактелию, врожденный порок сердца и ряд других аномалий (Рис. 39). В дальнейшем появились и другие описания синдрома. Помимо указанных аномалий, у больных детей обычно имеет место тяжелейшее поражение головного мозга, множественные пороки развития внутренних органов. Из внешних симптомов характерна флексорная контрактура пальцев и конечностей.
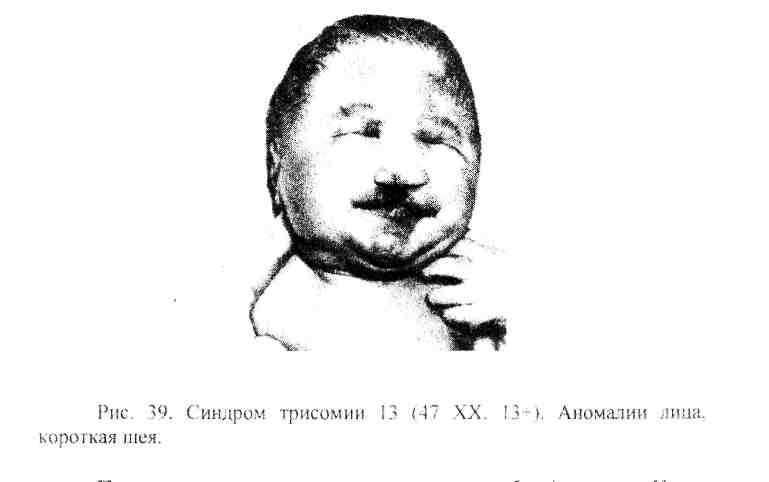
Продолжительность жизни не превышает 3-4 месяцев. Как и
при болезни Дауна могут быть случаи, обусловленные
транслокацией 13 хромосомы на другие хромосомы этой же
группы, значительно увеличивающие риск рождения больного
ребенка. В случае транслокации 13 на 13 риск 100%.
53.3. Синдром трисомии 18хромосомы
обычно у таких детей имеется дефект межжелудочковой перегородки и незаращение боталлова протока, соединяющего дугу аорты с легочными i артериями).

В дополнение к описанным дефектам можно добавить еще массу, однако достаточно ограничится обозначением их как множественные пороки развития органов. Как и при синдроме Патау, у больных отмечается флексорная деформация конечностей. В связи с тяжестью пороков развития около 70% детей погибают в первый месяц жизни.

Столь необычным название синдром обязан характерному крику, который издают до определенного возраста больные дети. По мнению одних авторов, это обусловлено аномалиями развития гортани, другие же полагают, что ыавная причина - поражение центральной нервной системы. Помимо своеобразного крика. болезнь характеризуется рядом других симптомов. У больных круглое лунообразное лицо, микроцефалия, микро- и ретрогнатия, гипертелоризм, антимонголоидный разрез глаз, эпикант (Рис. 41). низко расположенные диспластические уши, нередко четырехпалость, врожденные пороки сердца, задержка физического развития, умственная отсталость. Прогноз для жизни более благоприятен, нежели при двух предыдущих заболеваниях.
3.3.5. Моноеомия по X - хромосоме
Моносомия по X - хромосоме (синдром Шерешевского-Тернера. 45, X, рис. 40) впервые была описана в 1925 г. советским эндокринологом Н.А. Щерешевским, а в 1938 г.
Обычно причиной заболевания является отсутствие у плода одной из половых хромосом при сохранении в кариотипс X -хромосомы (кстати, у больных чаще, чем в среднем в популяции, встречается дальтонизм).

Предварительный диагноз можно поставить методом экспресс - диагностики путем исследования полового хроматина. При полной моносомии у женщины половой хроматин отсутствует. При неполной, обусловленной делецией одной из X - хромосом, половой хроматин будет присутствовать.
Описаны фенотипические проявления этого синдрома (низкий рост, перепончатая шея) и лиц мужского пола с нормальным для мужчин набором хромосом - 46, XY. Явления гипогонадизма отсутствуют.
Не нашли то, что искали? Воспользуйтесь поиском:
[youtube.player]Читайте также: