
Миопатия при ревматоидном артрите
Воспалительные миопатии — гетерогенная группа редких приобретенных заболеваний мышц. Характеризуется наличием воспалительного процесса в скелетных (поперечнополосатых) мышцах, вследствие которого происходят дегенеративные изменения. При воспалительных миопатиях воспаление носит аутоиммунный или инфекционный характер. Данный вид миопатии сопровождается:
- мышечной слабостью;
- болями в мышцах;
- атрофиями;
- наличием контрактур;
- понижением двигательной активности;
- возникновением отечности мышц;
- в некоторых случаях наблюдается мышечное уплотнение.
- Причины возникновения воспалительной миопатии
- Симптомы воспалительной миопатии
- Диагностика воспалительной миопатии
- Лечение воспалительной миопатии
Воспалительные миопатии — это большая группа заболеваний, включающая в себя системные поражения мышечной ткани (дерматомиозит, полимиозит, эозинофильный миозит, миозит с включениями, ревматическая полимиалгия), а также локальные воспаления в отдельных группах мышц (миозит глазных мышц, псевдотромбофлебит мышц голени и пр.).
Возраст людей, когда наиболее высок риск инфицирования воспалительными миопатиями, зависит от типа заболевания. Например, полимиозит наблюдается чаще после тридцати лет, миозит с включениями поражает людей старше сорока лет, являясь при этом самой распространённой формой воспалительных миопатий, встречающихся в пожилом возрасте.
Причины возникновения воспалительной миопатии
Причин возникновения воспалительных миопатий достаточно много. Врачи акцентируют внимание на том, что этология и патогенез заболеваний представленной группы до конца не изучены, не имеют четкой схемы.
- Некоторые инфекционные заболевания (грипп, ВИЧ-инфекции, краснуха и пр.) могут стать провокаторами воспалительной миопатии.
- Причинами, вызывающими воспалительные миопатии, могут стать также стрептококковые бактерии, паразитарные инвазии вроде токсоплазмоза или трихинеллёза, а также грибковые инфекции.
- Существуют также идиопатические миопатии (заболевания, возбуждение которых не связано с инфекциями), они имеют аутоиммунный механизм развития, но следует сказать, что антиген, который лежит в основе возбуждения аутоиммунной реакции, так и не вычислен. При изучении заболевания выявлены семейные случаи, исходя из чего можно судитьо генетической природе данной группы заболеваний.
К идиопатическим воспалительным миопатиям относят: полимиозит, миозит с включениями, миопатии при системных заболеваниях (системной красной волчанке, склеродермии, синдроме Шегрена, ревматоидном артрите, системных воскулитах). Пациенты с дерматомиозитом и полимиозитом чаще подвержены онкологическим заболеваниям, особенно в виде злокачественных опухолей.
Симптомы воспалительной миопатии
Воспалительные миопатии имеют следующую симптоматику:
- Болевой синдром в мышцах (миалгия). При прогрессировании болезни боли наблюдаются не только во время двигательной активности, но и в период покоя или при незначительном надавливании на мышцы.
- Мышечная слабость. Человек испытывает трудности при сжатии предметов в руках. Прогрессируя, заболевание делает почти невозможным поднятие руки или ноги, лишает больного возможности самостоятельно садиться и вставать.
- Значительное сокращение объема активных движений, а в отдельных случаях пациент приковывается к кровати.
- Отек и уплотнение пораженных мышц.
- Развитие атрофии и миофиброза (мышечные волокна замещаются на соединительнотканные).
- Кальциноз.
- Контрактуры.
- Тугоподвижность суставов.
- Миопатический парез гортани.
- Дыхательная недостаточность.
- Застойная и аспирационная пневмония (возникают как следствие слабости дыхательных мышц и снижения легочной вентиляции).
Клиническая картина заболеваний из группы воспалительных миопатий различна, как и продолжительность протекания. При дерматомиозите, например, симптомы начинают проявляться в виде общего недомогания и повышенной температуры, лихорадки. Впоследствии наблюдается характерная сыпь на кожных покровах, а затем слабость проксимальных мышц. Сыпь фиолетового цвета может наблюдаться на поверхностях век, сопровождается отечностью и телангиоэктазиями на шее и груди, между фалангами пальцев и на кисти — эритематозная сыпь. На суставах коленей и локтей появляется утолщение кожи. При дерматомиозите наблюдается гиперемия щек, дисколорация и утолщение ногтевых лож, что приводит к отечности пальцев. Далее, прогрессируя, болезнь приводит к тугоподвижности мышц и болевому синдрому. Происходит поражение в основном проксимальных мышц, дистальные мышцы менее поддаются травматизации.
У детей заболевание может проявлять себя как острый приступ или чередование обострений и ремиссий. При быстром прогрессировании может возникнуть дыхательная недостаточность, поскольку происходит поражение мышц гортани и дыхательных путей. У большей половины инфицированных детей были выявлены кальцификаты в подкожных тканях. В отдельных случаях болезнь заканчивается летально (около 10% всех инфицированных детей).
Почти у половины взрослых пациентов выявляются злокачественные опухоли при диагностике дерматомиозита (для сравнения у больных полимиозитом опухоли наблюдаются очень редко). В результате, симптоматика онкологического образования накладывается на клиническую картину дерматомиозита, что существенно усложняет диагностику и лечение заболевания. При удалении злокачественной опухоли происходит регресс мышечной слабости. Истинная причина злокачественных новообразований в клинической картине дерматомиозита остается неизвестной.
Диагностика воспалительной миопатии
Диагностика воспалительных миопатий затруднена. Это связано с широким разбросом клинической картины. Симптомы очень схожи с заболеваниями, которые относятся к группе воспалительных миопатий, а также заболеваниями иной природы. В процессе диагностики необходимо, в первую очередь, дифференцировать заболевание среди однородных ему. Пациент должен пройти консультации у невропатолога, дерматолога, пульмонолога и кардиолога для получения наиболее точной клинической картины и исключения похожих заболеваний.
При большинстве воспалительных миопатий (кроме миозита с включениями) может наблюдаться повышение СОЭ в крови, но почти у половины этот показатель показывает норму. Важным показателем при диагностике данного заболевания будет КФК (уровень креатинфосфокиназы), по которому можно судить о степени повреждения мышц и мышечной мембраны. Зачастую можно наблюдать повышения уровня изофермента КФК. Концентрация миоглобина в сыворотке тоже повышается и свидетельствует о явном прогрессировании заболевания и назначении адекватного лечения.
Диагностируя данное заболевания, назначают также ЭМГ, результаты которого важны для установления диагноза. При полимиозите и дерматомиозите, обычно наблюдаются кратковременные полифазные потенциалы двигательных единиц (это касается проксимальных мышц). Похожие изменения можно наблюдать также при миозите с включениями. Кроме того, похожие аномалии могут быть выявлены в процессе ЭМГ и при нейрогенном поражении. Поэтому установить точный диагноз, полагаясь лишь на ЭМГ, невозможно. Проводят также биопсию мышц. Этот анализ помогает определить распространённость воспалительного процесса.
Проводится гормональное исследование при отсутствии сыпи. Целью данного исследования будет определение содержания трийодтиронина, тиреотропного гормона TSH и тироксина.
Лечение воспалительной миопатии
Патогенического и этиологического лечения не существует. Лечение в основном направлено на устранение симптомов и общее поддержание мышечной силы, устранение контрактур, а также на достижение медикаментозной ремиссии.
Несмотря на ограниченное количество информации о способе лечения данных заболеваний, врачи пришли к единому мнению — иммуносупрессивная терапия дает хорошие результаты. Чаще всего пациенту назначается целый комплекс мероприятий. Во-первых, это медикаментозное терапевтическое лечение. Одной из самый успешных и эффективных методик является глюкокортикоидная терапия (преднизолон и метипред). Доза препаратов — 1 мг на 1 кг веса. Принимают препараты строго через день во избежание осложнений, которые могут появиться вследствие применения глюкокортикоидной терапии. Назначая такую терапию, врач должен тщательно обследовать пациента на наличие сопутствующих заболеваний, которые могут вызвать осложнения. К таким заболеваниям, при которых глюкокортикоидная терапия противопоказана относятся:
- сахарный диабет;
- остеопороз;
- язва желудка;
- ожирение или любые другие нарушения обмена веществ;
- повышенное артериальное давление.
Ремиссия наступает приблизительно через один-два месяца. После этого доза препаратов уменьшается под четким контролем игольчатой ЭМГ. Иногда лечащим врачом принимается решение о замене базового препарата при условии значительных улучшений в анализе клинической картины. Чаще всего препаратом-заменителем выступает азатиоприн. Если глюкокортикоидная терапия не дала желаемых результатов, то для усиления эффекта параллельно назначается цитостатическая терапия.
Для скорейшей ремиссии врачи советуют также лечебную физкультуру (строго под присмотром методиста по ЛФК или компетентного специалиста), проведение курса массажей, различные ортопедические мероприятия. Врачи рекомендуют активное движение, длительное лежание противопоказано. Преждевременное использование кресла-каталки также не рекомендуется, поскольку предусматривает покой значительной части мышц.
Говоря о прогнозах данной группы заболеваний, то они неплохие. При своевременном и адекватном лечении около половины больных существенно улучшают свое общее состояние и полностью излечиваются. Треть пациентов остается с ощущением мышечной слабости. В течение первых трех-четырех лет примерно 15% пациентов с тяжёлой формой умирают от сопутствующих заболеваний сердца, легких и пр. Значительная часть смертельных случаев происходит по причине образовавшихся онкологических опухолей.
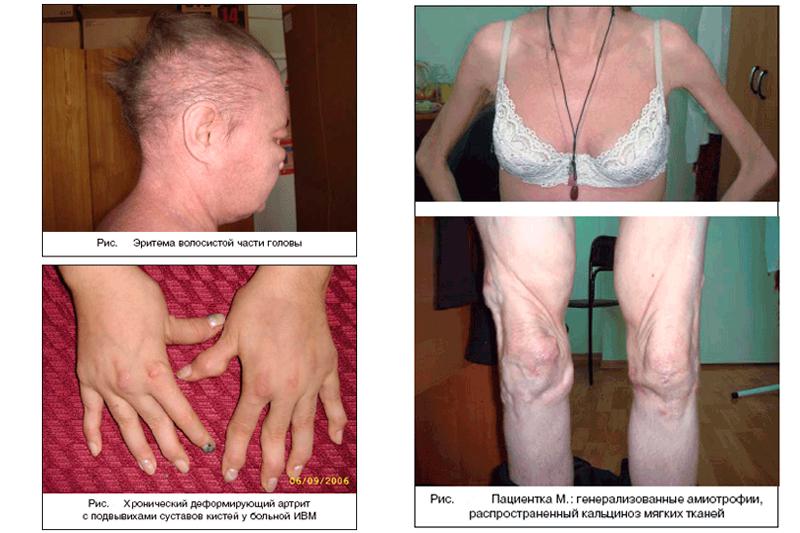
Мышцы поражаются у большинства больных РА. Мышечная слабость и атрофия отмечаются не менее чем у 75% больных. Причиной этих симптомов служит уменьшение мышечной активности в связи с болью в суставах и особенно их контрактурами. Эта причина, однако, не может считаться единственной. Еще в 1873 г. J. Paget писал, что мышечная атрофия при РА пропорциональна не столько ограничению движений, сколько выраженности боли.
Спустя почти столетие J. De Andrade (1968) доказал в эксперименте, что атрофия мышц у животных с артритами выражена сильнее, чем вызванная иммобилизацией. О том же свидетельствуют частые клинические наблюдения, касающиеся быстро развивающейся амиотрофии при тяжелом РА. Темпы ее развития при этом более быстрые, чем при обычной иммобилизации, тем более что в подобных случаях общий объем движений часто остается немалым.
Поэтому основными причинами мышечной патологии у больных РА следует считать как ограничение мускульной активности, так и катаболическое влияние воспалительного процесса (возможно, опосредуемое эффектом интерлейкина-1 и фактором некроза опухоли, вырабатываемых макрофагами). Последнее может значительно усугубляться сопутствующей лихорадкой, частым ухудшением аппетита, нерациональным питанием (в частности, бедным белками).
Трактовка данных биопсий вне связи с клиническими и биохимическими показателями должна быть очень осторожной. Микроскопический анализ мышечной ткани нередко обнаруживает сосудистые изменения и клеточные инфильтраты, формально соответствующие представлениям о воспалении, у лиц без какой-либо мышечной патологии. Возможно, что речь идет о реакции на транзиторные физиологические сдвиги (большее накопление молочной кислоты и т. д.).
Не подтвердилось также мнение об относительной специфичности для РА так называемого узелкового полимиозита, представляющего собой узелковые клеточные инфильтраты диаметром 1—2 мм, обильно рассеянные в эндомизии и перимизии и состоящие из скоплений лимфоцитов и плазматических клеток, в редких случаях — тучных клеток, нейтрофилов и эозинофилов. L. Sokoloff и соавт. (1950) показали, что подобные изменения наблюдались у 56% больных РА, у 29% больных неревматическими заболеваниями и у 23% здоровых.
Повышение уровня мышечных ферментов в крови имеет большое значение для диагноза ревматоидного миозита. Однако необходимо принимать во внимание, что содержание этих ферментов у больных РА может повыситься также вследствие внутримышечных инъекций, мышечной биопсии, индивидуальных реакций на некоторые лекарства (хлорохин, ацетилсалициловая кислота, D-пеницилламин) и даже значительной физической нагрузки. В связи с этим диагностика ревматоидного миозита надежна лишь при сочетании клинических симптомов с ферментемией и гистологическими воспалительными изменениями или (что менее убедительно) при комбинировании двух компонентов этой триады.
Четко очерченный синдром миозита у больных РА редок. Относительно чаще он встречается при ревматоидном васкулите, однако и в этих случаях (при локализации васкулита в мышцах) его клиническое значение обычно невелико. Поэтому особый интерес вызывает наблюдение Г. Н. Жуковской (1987) больного классическим серопозитивным и эрозивным РА (без васкулита), у которого миозит в течение длительного времени настолько превалировал над суставными проявлениями, что правильный диагноз был поставлен более чем через год.
Необходимо иметь в виду, что заметная мышечная патология у больных РА в редких случаях может быть связана с влиянием некоторых лекарств. Так, глюкокортикостероиды (прежде всего содержащие фтор — триамцинолон, дексаметазон), особенно при их длительном назначении в больших дозах, вызывают у отдельных больных так называемую стероидную миопатию. Ее симптомы обычно развиваются постепенно и относятся преимущественно к проксимальным мышцам ног, а затем и рук (боли, быстрая утомляемость, затруднения при подъеме на лестницу и при вставании с низкого стула).
Эти симптомы нередко оцениваются как признаки обострения основного заболевания, что ведет к увеличению дозы стероидов и тем самым к усугублению миопатических проявлений. Уровень сывороточных мышечных ферментов при стероидной миопатии нормален. Ценный лабораторный признак — повышенное выделение креатина с мочой. При гистологическом исследовании мышц не обнаруживают достоверных воспалительных изменений; отмечаются лишь неспецифические дистрофические симптомы — потеря поперечной исчерченности, атрофия волокон и т. д. После уменьшения дозы гормональных препаратов и особенно полной отмены их признаки стероидной миопатии исчезают, чему способствует лечебная физкультура, направленная на упражнения для соответствующих мышц.
Назначение D-пеницилламина у некоторых больных может вызвать патологию мышц двух различных типов. Относительно чаще встречается синдром миастении (myasthenia gravis). Он проявляется в постепенном нарастании слабости различных мышечных групп с соответствующими функциональными нарушениями (слабость, утомляемость, диплопия, нарушения глотания и речи, птоз, затруднения при выполнении простых движений).
Повышения уровня мышечных ферментов в крови и гистологической картины миозита не наблюдается. У некоторых больных . определяются антитела к тканевым компонентам мышечных поперечных полос и к ацетилхолиновым рецепторам мышц (последнее обстоятельство может быть непосредственной причиной мышечной слабости). После отмены D-пеницилламина симптомы миастении постепенно исчезают; при недостаточном восстановлении мышечной силы назначают прозерин.
У единичных больных при назначении D-пеницилламина описано развитие истинного полимиозита с миалгиями, слабостью проксимальных мышц конечностей, нарастанием уровня мышечных ферментов и характерной гистологической картиной (инфильтрация лимфоцитами и макрофагами, участки дегенерации и регенерации мышечных волокон). Для лечения этого осложнения необходимо отменить D-пеницилламин, в серьезных случаях — назначить глюкокортикостероиды.
Хлорохиновая миопатия также представляет собой очень редкое лекарственное осложнение. Она характеризуется постепенно прогрессирующей слабостью проксимальных мышц ног, затем — рук, позже — более дистальных мышечных групп. Уровень мышечных ферментов в крови может быть повышен. Характерный гистологический признак — вакуолизация мышечных волокон. Отмена хлорохина приводит к полному, но медленному выздоровлению.
Назначение гидроксихлорохина (плаквенила) не вызывает миопатии. Таким образом, в основе изменений мышц при РА могут лежать принципиально различные причины: миозит, атрофия вследствие недостаточных упражнений, вызванные воспалительным процессом катаболические сдвиги в обмене веществ, недостаточное белковое питание и, наконец, редкие и сугубо индивидуальные (возможно, генетически обусловленные) реакции на определенные лекарственные препараты.
Поэтому в каждом конкретном случае явной мышечной патологии у больного РА необходим соответствующий клинический и анамнестический анализ с определением в крови уровня мышечных ферментов; по показаниям проводят биопсию наиболее пораженных отделов мышц.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Основные формы миопатии:
- I. Наследственные прогрессирующие мышечные дистрофии: Мышечная дистрофия Дюшенна и Беккера (Duchenne-strongecker), дистрофия Эмери-Дрейфуса (Emery-Dreifuss), фацио-скапуло-хумеральная, скапулоперонеальная, конечностно-поясная, дистальная форма, окулофарингеальная, прогрессирующая наружная офтальмоплегия. Конгенитальная мышечная дистрофия.
- II. Миопатии с миотоническим синдромом (мембранные миопатии).
- III. Воспалительные миопатии: полимиозит, СПИД, коллагенозы и др.
- IV. Метаболические миопатии (в том числе эндокринные и митохондриальные миопатии; миоглобулинемия и др.).
- V. Ятрогенные и токсические миопатии.
- VI. Алкогольная миопатия.
- VII. Паранеопластическая миопатия.

[1], [2], [3], [4], [5], [6]
Код по МКБ-10
Где болит?
Мышечные дистрофии - термин, который применяют для обозначения наследственных форм миопатии, сопровождающихся дегенерацией мышц. Это - целая группа заболеваний, большинство из которых начинается в детском или юношеском возрасте, имеет неуклонно прогрессирующее течение и рано или поздно приводит к тяжёлой инвалидизации. Предложено несколько подробных классификаций мышечных дистрофий, построенных на разных принципах (генетических, биохимических, клинических), но унифицированнной классификации нет.
Дистрофин-дефицитные дистрофии объединяют в основном две формы: мышечную дистрофию Дюшенна (Duchenn) и мышечную дистрофию Беккера (strongecker).
Мышечная дистрофия Дюшенна, или псевдогипертрофическая мышечная дистрофия Дюшенна - самая злокачественная и самая распространённая форма Х-сцепленных миодистрофии. ферментемия (КФК) выявляется уже в неонатальном периоде, но клинические симптомы появляются в возрасте 2-4 лет. Эти дети поздно начинают ходить, им трудно или невозможно бегать и прыгать, они часто падают (особенно при попытке бежать), с трудом поднимаются по ступенькам или по наклонному полу (слабость проксимальных мышц) и ходят на больших пальцах (toe-walking) из-за контрактуры сухожилий стопы. Возможно снижение интеллекта. Характерна псевдогипертрофия икроножных мышц. Постепенно процесс принимает восходящее направление. Формируется гиперлордоз и кифосколиоз. К 8-10 годам грубо нарушается походка. Больной встаёт с пола с помощью характерных «миопатических «приёмов. К 14 - 15 годам больные, как правило, полностью обездвижены и умирают к 15-17 годам от слабости дыхательных мышц грудной клетки. ЭКГ обнаруживает нарушения почти в 90 % случаев (кардиомиопатия). Уровень КФК резко повышен. На ЭМГ - мышечный уровень поражения. В биоптате мышц неспецифические, хотя и характерные гистопатологические нарушения.
Мышечная дистрофия Беккера - вторая по частоте, но доброкачественная форма псевдогипертрофической миодистрофии. Начало заболевания между 5 и 15 годами. Паттерн вовлечения мышц такой же как при форме Дюшенна. Характерна слабость мышц тазового пояса и проксимальных отделов ног. Изменяется походка, появляются затруднения при вставании с низкого стула, при подъёме по лестнице. Развивается выраженная псевдогипертрофия икроножных мышц; процесс медленно распространяется вверх на мышцы плечевого пояса и проксимальных отделов рук. Уровень КФК повышен
Течение заболевание более благоприятное и медленное с более поздним нарушением трудоспособности.
Окулофарингеальная мышечная дистрофия характеризуется поздним началом (на 4-6 декаде жизни) и проявляется поражением глазодвигательных мышц, а также мышц глотки с нарушением глотания. Существует и форма с изолированным поражением только глазодвигательных мышц, которая, постепенно прогрессируя, приводит в конце концов к полной наружной офтальмоплегии. Последняя обычно протекает без двоения (окулярная миопатия, или прогрессирующая наружная офтальмоплегия Грефе). Диагноз подтверждается ЭМГ-исследованием. Уровень КФК поднимается редко (если процесс распространяется на другие поперечнополосатые мышцы).
Скапулоперонеальная (лопаточно-перонеальная) амиогрофия Давиденкова характеризуется прогрессирующей атрофией и слабостью в перонеальных группах мышц, а затем и в мышцах плечевого пояса. Некоторые исследователи считают, что синдром лопаточно-перонеальной атрофии является вариантом развития миодистрофий Ландузи-Дежерина.
Дистальная мышечная дистрофия является исключением из всей группы миодистрофий, так как поражает дистальную мускулатуру сначала голеней и стоп, затем - рук. Сухожильные рефлексы выпадают в той же последовательности Редко процесс распространяется на проксимальную мускулатуру. Для диагноза необходима сохранность чувствительности и нормальная скорость проведения возбуждения по нервам. Уровень КФК нормальный или слабо повышенный. ЭМГ подтверждает мышечный уровень поражения.
Существуют варианты дистальной мышечной дистрофии с началом в младенчестве, в детском возрасте, с поздним началом (тип Веландер), с накоплением десминовых включений.
Мышечная дистрофия Эмери-Дрейфуса имеет Х-сцепленный тип наследования, начинается в 4-5-летнем возрасте с характерным плече-перонеальным распределением атрофии и слабости (дистальные отделы остаются сохранными даже в далеко зашедших случаях). Типично раннее формирование контрактур в локтевых суставах, шее и в области ахилловых сухожилий. Другая типичная особенность - отсутствие псевдогипертрофий. Характерны нарушения ритма сердца, дефекты проведения (иногда полная блокада с внезапной смертью больного). Уровень КФК в сыворотке остаётся долгое время нормальным. ЭМГ показывает как нейрогенный, так и мышечный уровень поражения.
Особая группа - конгенитальные миопатии объединяет несколько заболеваний, которые выявляются обычно с рождения или в раннем детском возрасте и отличаются доброкачественным течением: часто они остаются стабильными на протяжении всей жизни; иногда они даже начинают регрессировать; если же отмечается в ряде случаев прогрессирование, то оно очень незначительное.
ЭМГ выявляет при этих формах неспецифические миопатические изменения. Мышечные фермены в крови либо нормальные либо слегка повышены. Диагноз ставится на основании электроно-микроскопического исследования.
Мембранные миопатии
Так называемые мембранные миопатии, куда относятся миотонические синдромы.
Воспалительные миопатии
В группу воспалительных миопатий включены такие заболевания как полиомиозит и дерматомиозит; миозит и миопатия с тельцами включения; миозит при заболеваниях соединительной ткани; саркоидная миопатия; миозит при инфекционных заболеваниях.
Наличие кожных изменений (эритемы, нарушения пигментации, телеангиоэктазии) является главным отличием дерматомиозита от полимиозита. Полиомиозит может быть первичным и вторичным (при злокачественном новообразовании).
Поражает чаще больных среднего или пожилого возраста (преобладают мужчины) и проявляется медленно прогрессирующей симметричной слабостью в конечностях. В отличие от других воспалительных миопатии здесь характерна как проксимальная, так и дистальная выраженная слабость мышц с вовлечением экстензоров стопы и флексоров пальцев рук. Боль не характерна. Иногда миозит с тельцами включения сочетается с заболеваниями соединительной ткани или иммунными нарушениями (болезнь Шегрена, тромбоцитопения). Уровень КФК умеренно повышен. ЭМГ обнаруживает смешанные нейрогенные и миопатические изменения в характере биоэлектрической активности. Биопсия мышц выявляет маленькие вакуоли с гранулами включения.
Такое сочетание особенно характерно для случаев смешанного (mixed) соединительнотканного заболевания. Оно характеризуется высокими титрами антирибонуклеопротеиновых антител; волчаночно-подобными высыпаниями на коже; изменениями соединительной ткани, напоминающими склеродермию; артритом и воспалительной миопатией. Клинически миопатия проявляется слабостью сгибателей шеи и мышц проксимальных отделов конечностей. Гистологически эта воспалительная миопатия напоминает дерматомиозит.
Воспалительная миопатия может наблюдаться при склеродермии, ревматоидном полиартрите, системной красной волчанке, синдроме Шегрена.
Может наблюдаться при саркаидозе (мультисистемное грануломатозное расстройство неизвестной этиологии). Грануломатозные изменения находят в оболочках мозга, головном мозге, гипофизе, спинном мозге и периферических нервах (а также в тканях глаза, в коже, лёгких, костях, лимфатических узлах и слюнных железах) Диагноз ставится на основания выявления мультисистемного поражения и биопсии мышц.
Бактериальные и грибковые миозиты встречаются редко и обычно являются компонентом системного заболевания. Паразитарные миозиты (токсоплазмоз, трихинеллёз, цистицеркоз) также встречаются нечасто. При цистицеркозе описана псевдогипертрофическая миопатия. Вирусные миозиты могут проявляться разной степенью тяжести от миалгии до рабдомиолиза. Разновидность таких воспалительных миопатии характерна для осложнений ВИЧ-инфекции и обычно наблюдается в контексте других неврологических и соматических проявлений СПИДа.
Метаболические миопатии
Метаболические миопатии включают карбогидратные миопатии, липидные миопатии, митохондриальные миопатии, эндокринные миопатии, миалгические синдромы, миоглобулинурию и токсические миопатии.
Карбогидратные миопатии обозначают как болезни накопления гликогена. Они связаны с недостаточностью тех или иных ферментов. Недостаточность мышечной фосфорилазы (болезнь Мак-Ардла) и других ферментов, а также липидные миопатии. Среди этих заболеваний осталась неупомянутой лизосомная болезнь накопления гликогена (болезнь Помпе - Ротре), проявляющаяся в первые месяцы жизни (быстро прогрессирующая мышечная слабость и массивная кардиомегалия) и приводящая к смерти на первом году жизни.
Синдром Кирнса-Сейра (Kearns-Sayre) проявляется прогрессирующей наружной офтальмоплегией. Он относится к спорадическим заболеваниям (но существует и семейный вариант прогрессирующей наружной офтальмоплегии) и, что типично, сопровождается вовлечением многих органов и систем. Болезнь начинается до 20-летнего возраста и проявляется пигментной дегенерацией сетчатки. Облигатные признаки этого заболевания: наружная офтальмоплегия, нарушения проводимости сердца и упомянутая пигментная дегенерация сетчатки. В качестве других дополнительных симптомов отмечают атаксию, ухудшение слуха, множественную эндокринопатию, увеличение содержание белка в ликворе и другие проявления. При семейном варианте прогрессирующей наружной офтальмоплегии возможны проявления слабости в мышцах шеи и конечностей.
Эндокринные миопатии встречаются при самых разнообразных эндокринных расстройствах. Довольно часто миопатия наблюдается при гипертиреозе. Слабость выявляется преимущественно в проксимальных отделах конечностей (редко - в дистальной и бульбарной мускулатуре) и подвергается обратному развитию при лечении гипертиреоза. Уровень КФК обычно не повышен. На ЭМГ и в мышечном биоптате - неспецифические миопатические изменения.
Однако встречаются случаи выраженного тиреотоксикоза, особенно при его быстром прогрессировании, сопровождающиеся рабдомиолизом, миоглобинурией и почечной недостаточноcтью. Слабость респираторых мышц, требующая механической вентиляции встречается редко.
Гипотиреоз часто сопровождается проксимальной мышечной слабостью, крампи, болью и ощущением напряжения (stiffness) мышц (хотя при объективном измерении слабость подтверждается нечасто). Эти симптомы исчезают при успешном лечении гипотиреоза. Мышечные гипертрофии редко встречаются при гипотиреозе, но их наличие у взрослых носит название синдрома Гофмана.
Кохер-Дебре-Семелайгна синдром наблюдается у детей (гипотиреоз с генерализованным мышечным напряжением и гипертрофией икроножных мышц). Уровень КФК повышен у 90 % больных гипотиреозом, хотя явный рабдомиолиз наблюдается очень редко. Миопатические изменения на ЭМГ вариируют от 8 % до 70 %. В мышечном биоптате - слабо выраженные признаки миопатии. Гипотиреоз ухудшает гликогенолиз в мышцах и окислительную способность митохондрий.
Мы не обсуждаем здесь дистиреоидную орбитопатию, также связанную с поражением мышечного аппарата орбиты.
Мышечная слабость, утомляемость и крампи очень часто сопутствуют болезни Аддисона. Иногда слабость может быть эпизодической. Здесь может иметь место периодический паралич с квадриплегией и гиперкалиемией.
Больные с гиперальдостеронизмом иногда отмечают приступы периодического паралича с гипокалиемией. 70 % этих больных жалуются на слабость.
На мышечную слабость часто жалуются больные с синдромом Иценко-Кушинга и больные, получающие длительное лечение глюкокортикоидами. Стреоидная миопатия чаще развивается медленно при длительном лечении таких, например, заболеваний, как системная красная волчанка, ревматоидный артрит, бронхиальная астма, полимиозит, и поражает преимущественно проксимально расположенные мышцы. Уровень КФК обычно не меняется; на ЭМГ - минимальные признаки миопатии.
Острая стероидная миопатия развивается реже: часто спустя неделю от начала лечения высокими дозами кортикостероидами. Такая миопатия может вовлекать респираторные мышцы. Острая стероидная миопатия может возникать и при лечении кортикростероидами больных миастенией.
Токсические миопатии
Токсические миопатии могу носить ятрогенный характер. Лекарственные препараты способны вызывать: миалгии, напряжение мышц (stiffness), или крампи; миотонию (задержку релаксации скелетных мышц после произвольного сокращения) - безболезненную проксимальную миопатию с мышечной слабостью; миозит или воспалительную миопатию; фокальную миопатию в области повреждения (инъекции); гипокалиемическую мимопатию при введении лекарств, вызывыающих гипокалиемию; митохондриальную миопатию в связи с торможением митохондриальной ДНК; рабдомиолиз (острый мышечный некроз с миоглобинурией и системными осложнениями).
Некротизирующая миопатия описана при использовании ловастатина (ингибитора синтеза холестерола), циклоспорина, аминокапроновой кислоты, прокаинамида, фенциклидина. Развивается мышечная слабость, боли (спонтанные и при пальпации мышц); уровень КФК повышается; на ЭМГ - картина миопатических изменений. Внутримышечное введение антибиотиков доксина ботулизма, хлорпромазина, фенитиона, лидокаина и диазепама может быть причиной локального мышечного некроза и фиброзной миопатии. Эметин вызывает прогрессирующую проксимальную миопатию. Такой же способностью обнаружена у клозапина, D-пеницилламина, гормона роста, интерферон-альфа-2b, винкристина.
Миалгию и мышечные крампи могут вызвать: ингибиторы ангиотензин-конвертирующего фактора, антихолинестераза, бета-адренергические агонисты, антагонисты кальция, отменам кортикостероидов, цитотоксические препараты, дексаметазон, диуретики, D-пеницилламин, левамизол, литий, L-триптофан, нифедипин, пиндолол, прокаинамид, рифампицин, сальбутамол. Лекарственно-индуцированная миалгия без мышечной слабости обычно быстро проходит после отмены препарата.
Алкогольная миопатия
Встречается в нескольких вариантах. Один тип характеризуется безболезненной, преимущественно проксимальной мышечной слабостью, развивающейся в течение нескольких дней или недель длительного злоупотребления алкоголем, что сочетается с выраженной гипокалиемией. Уровень печёночных и мышечных ферментов заметно повышается.
Другой тип алкогольной миопатии развивается остро на фоне длительного употребления алкоголя и проявляется выраженной болью и отёчностью мышц конечностей и туловища, что сопровождается симптомами почечной недостаточности и гиперкалиемией. Мионекроз (рабдомиолиз) отражается в высоком уровне КФК и альдолазы, а также миоглобинурией. Этому могут сопутствовать другие синдромы алкоголизма. Восстановление довольно медленное (недели и месяцы); характерны рецидивы, связанные с алкоголизмом.
Встречается вариант острой алкогольной миопатии, сопровождающийся тяжёлыми крампи и генерализованной слабостью. Возможна хроническая алкогольная миопатия, проявляющаяся безболезненной атрофией и слабостью мышц проксимальных отделов конечностей, особенно ног, с минимально выраженными знаками нейропатии.
[7], [8], [9], [10], [11]
Паранеопластическая миопатия
Отдельную позицию должна занимать миопатия с остеодистрофией и остеомаляцией, которая описана среди прочих паранеопластических синдромов.
Здесь не представлены некоторые редкие формы мышечных дистрофий, такик как миодистрофия Мэбри, миодистрофия Роттауфа-Мортье-Бейера, тазово-бедренная миодистрофия Лейдена-Мёбиуса, мышечная дистрофия Бетлема, дистальная миодистрофия Миоши.
Диагностика миопатий
Диагностические исследования при подозрении на миопатию включают, помимо клинического анализа, электромиоргафическое и электронейромиографическое исследование, анализ крови на ферменты (КФК, альдолаза, ACT, АЛТ, ЛДГ и др.). КФК в крови - наиболее чувствительный и достоверный показатель миодистрофического процесса. Исследуют также мочу на креатин и креатинин. Мышечная биопсия иногда незаменима для выявления природы миопатии (например, при конгенитальных миопатиях). Точная диагностика типа миопатии может потребовать проведения молекулярно-генетических, иммунобиохимических или иммуногистохимических исследований.
[12], [13], [14], [15], [16]
Читайте также: