
Наследственные мышечные заболевания мальчиков
Спинальная мышечная атрофия относится к генетически обусловленным заболеваниям и характеризуется прогрессирующей атрофией мышц в результате нарушения периферической иннервации. При этом происходит блокада импульсов спинного мозга, идущих к мышечным волокнам, что вызывает прекращение их нормального развития и функционирования. Чаще поражаются мышцы туловища и проксимальных отделов конечностей. Болезнь развивается у новорожденных детей, но иногда поражает людей молодого возраста. 
Причины развития болезни
Развитие патологии связывают с мутацией генов, которая приводит к нарушению синтеза специфического белка, участвующего в жизнедеятельности двигательных нейронов спинного мозга. Они представляют собой скопление клеток, которые локализуются в передних спинномозговых отделах и образуют двигательные корешки. Главной функцией нейронов считается регулирование сократительной активности, кровообращения, обменных процессов мышечных волокон.
При нарушении синтеза белка мотонейроны постепенно разрушаются как в правой, так и левой половине спинного мозга. Симметричная спинная мышечная атрофия относится к важному диагностическому признаку.
В результате к мышцам не поступают сигналы из центральной нервной системы, они теряют способность к сокращению. Это негативно влияет на двигательную активность и приобретение ребенком навыков самообслуживания. При этом чувствительная иннервация сохраняется в полном объеме.
Заболевание передается по аутосомно-рецессивному типу, поэтому встречается в популяции населения крайне редко. Для того чтобы патология начала развиваться в организме, необходимо носительство дефектного гена обоими родителями. При этом чаще всего у них нет проявлений заболевания, а обоюдное носительство не всегда дает появление недуга у совместного ребенка. Доказано, что каждый 40 человек может содержать в своем генетическом материале мутации определенного вида хромосом, обуславливающие носительство дефектного гена.
Проявления заболевания
Выделяют несколько форм развития патологии, которые отличаются возрастом возникновения первых симптомов, тяжестью течения, продолжительностью жизни.
Чем позже развивается патология, тем лучше прогноз для жизни и поддержания навыков самообслуживания.
В любом случае больные становятся инвалидами, требующими дополнительный уход и специальных средств передвижения (коляски, ходунки). Часто пациенты прикованы к кровати, их состояние осложняется застойными процессами в легких, которые могут привести к летальному исходу.

Спинальная мышечная атрофия тип 1 может быть заподозрена еще во внутриутробном периоде при слабом шевелении ребенка во второй половине беременности. На протяжении полугода после рождения отмечают вялость, низкую активность новорожденного. При обследовании не обнаруживают сухожильные рефлексы (ахиллов, коленный).
Мышцы верхних и нижних конечностей, туловища и головы не развиваются. Слабость и атрофия мышечных волокон приводит к неспособности детей полноценно сидеть, ползать, вставать на ноги. По мере роста скелета возникает деформация костей. Например, при попытке новорожденного сидеть позвоночник не поддерживается мышечным каркасом спины, что приводит к развитию кифоза.
Нередко выявляют непроизвольное подергивание мышц, дрожание пальцев вытянутых рук. Мышечная атрофия может визуально сглаживаться чрезмерным развитием подкожно-жировой клетчатки. Из-за недоразвития межреберных мышц грудная клетка приобретает уплощенную форму. Страдает функция глотания, сосания, дыхания, в результате чего развиваются тяжелые осложнения: аспирационная и застойная пневмония, истощение, дыхательная недостаточность.
Развитие умственной сферы проходит удовлетворительно, так как отделы головного мозга не подвергаются патологическим изменениям.
Эта форма заболевания считается неблагоприятной для жизни, такие дети обычно погибают в течение первых лет после рождения.
Атрофия спинного мозга в случае 2 типа болезни проявляется в конце первого года жизни, обычно в 7−18 месяцев ребенка. Признаки патологии выражены не резко и развиваются постепенно. По мере взросления новорожденных отмечают слабую двигательную активность. Дети предпочитают лежать, неохотно перемещаются в пространстве, значительно отстают в физическом развитии от своих сверстников.
Мышцы тела и конечностей подвержены непроизвольным сокращениям, отмечают подергивание языка. Дети способны сидеть, вставать на ноги, иногда медленно передвигаться с посторонней помощью. Прием пищи и дыхание обычно не затруднено, сухожильные рефлексы развиты слабо. Продолжительность жизни таких больных снижают частые респираторные инфекции и тяжелые застойные воспаления легких в результате вялой двигательной активности.
Первые симптомы недуга появляются в конце второго года жизни ребенка, иногда развитие мышечной атрофии начинается в период полового созревания. Заболевание характеризуется медленным прогрессированием. Сначала атрофируются верхние мышечные группы ног, затем патологический процесс поражает руки, туловище, шею. 
Больные, страдающие 3 типом болезни, способны передвигаться: вставать, ходить, подниматься по лестнице. Характерным признаком заболевания считается гипертрофия ягодичных и икроножных мышц. Однако по мере нарастания дистрофических изменений в передних отделах спинного мозга сухожильные рефлексы угасают, что свидетельствует о необратимых изменениях мышечной ткани.
Со временем двигательная активность снижается, что заставляет больных пользоваться инвалидными колясками. Длительно сохраняется способность к самообслуживанию, тазовые функции развиты хорошо (мочеиспускание, дефекация). Чувствительная иннервация при заболевании не страдает.
Терапевтическая тактика
Лечение спинальной мышечной атрофии имеет поддерживающий характер и проводится в течение всей жизни больного.
Выздоровления достичь невозможно, а лишь замедлить развитие патологии в нервной и мышечной ткани.
Терапевтическая тактика включает комплекс мер, направленных на сохранении двигательной активности.
- Лекарственные средства, нормализующие проведение нервного импульса — прозерин, нивалин.
- Ноотропные препараты, улучшающие кровообращение нервной ткани — пирацетам, ноотропил.
- Средства, стимулирующие кровоток и обменные реакции в мышечной ткани — актовегин, оротат калия.
- Витаминные препараты, обладающие антиоксидантным действием — токоферол, витамины группы В, глутаминовая кислота.
- Физиопроцедуры, усиливающие кровообращения пораженных конечностей — парафин, УВЧ, электрофорез с прозерином.
- Массаж — стимулирует мышечную активность, способствует выведению продуктов обмена.
- Лечебная гимнастика — атрофия мышц спины, рук, ног после занятий замедляется.
- Ортопедическая помощь — использование специальных приспособлений, поддерживающих двигательную активность грудной клетки, верхних и нижних конечностей.
В терминальных стадиях заболевания появляются дыхательные расстройства, которые требуют перевода пациентов на искусственную вентиляцию легких.
Спинальная мышечная атрофия считается тяжелым генетическим заболеванием с постоянным прогрессированием патологического процесса и неблагоприятным прогнозом.
В последние годы ученые всего мира ведут научные разработки по синтезированию лекарственного препарата, способного замещать недостающий нейронный белок. Эта единственная надежда больных на выздоровление и значительное улучшение качества жизни.
Существует огромное количество различных заболеваний, которые возникают у деток независимо от обстоятельств или действия окружающей среды. Это категория именно наследственных болезней. Сейчас же пойдет речь о такой проблеме, как мышечная дистрофия Дюшенна: что это за хворь такая, каковы у нее симптомы и можно ли с ней справиться.
Терминология
Изначально нужно узнать, что же такое наследственные болезни. Так, это заболевания, которые возникают в результате дефектов аппарата наследственных клеток. То есть это определенные сбои, которые происходят на генетическом уровне.
Мышечная дистрофия Дюшенна – это именно наследственная болезнь. Проявляется она очень быстро, основной симптом в данном случае – это быстро прогрессирующая слабость в мышцах. Нужно отметить: как и все остальные мышечные заболевания, болезнь Дюшенна также приводит в конечном результате к атрофии мышц, нарушению моторики и, конечно же, инвалидности. В подростковом возрасте детки с таким диагнозом уже не имеют возможности самостоятельно передвигаться и не могут обходиться без посторонней помощи.
Что происходит на генном уровне
Как уже было отмечено, мышечная дистрофия Дюшенна – это генетическое заболевание. Так, происходит мутация в том гене, который отвечает за выработку особого белка дистрофина. Именно он и необходим для нормальной работы мышечных волокон. При этом важно отметить, что эта генетическая мутация может как передаваться по наследству, так и возникать спонтанно.
Также важно отметить, что ген локализируется в хромосоме Х. Но женщины этой болезнью заболеть не могут, являясь только лишь передатчиком мутации от поколения к поколению. То есть если мама передаст мутацию сыну, он с 50%-й вероятностью заболеет. Если же девочке, она просто будет носителем гена, клинических проявлений болезни у нее не будет.
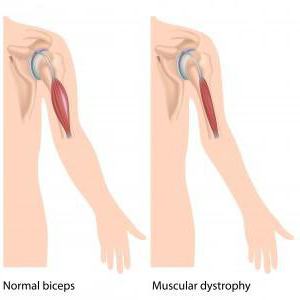
Симптоматика: группы
В основном, болезнь активно заявляет о себе примерно в 5-6 летнем возрасте. Однако первые симптомы могут возникнуть у малыша, который еще не достиг трехлетнего возраста. При этом надо отметить, что все патологические нарушения медки условно разделяют на несколько больших групп:
- Поражение мускулатуры.
- Поражение сердечной мышцы.
- Деформация скелета ребенка.
- Различные эндокринные расстройства.
- Нарушения нормальной умственной деятельности.
Наиболее часто встречающиеся проявления болезни
Обязательно также надо рассказать о том, как проявляется синдром Дюшенна. Симптомы бывают следующие:
- Слабость. Которая постепенно нарастает, развивается.
- Начинается прогрессирующая мышечная слабость именно с верхних конечностей, далее затрагиваются ноги и только потом – все остальные части тела и органы.
- Ребенок утрачивает возможность сам передвигаться. Примерно к 12-летнему возрасту такие детки уже полностью зависимы от инвалидной коляски.
- Также наблюдаются расстройства дыхательной системы.
- Ну и, конечно же, бывают нарушения в работе кардиологической системы. Позже происходят необратимые изменения в миокарде.
О поражении мышц скелета
Именно поражения мышечной ткани – наиболее распространенный симптом, если речь идет о такой проблеме, как синдром Дюшенна. При этом надо отметить, что рождаются детки без особых отклонений в развитии. В малом возрасте ребята менее активны и подвижны, нежели сверстники. Но чаще всего это связывают с темпераментом и характером ребенка. Поэтому отклонения очень редко замечаются. Более существенные признаки проявляются уже во время ходьбы малыша. Такие детки могут передвигаться на носочках, не становясь на полную стопу. Также они частенько падают.
Особым показателем является еще и симптом Говерса. То есть ребенок, чтобы подняться с пола, активно пользуется руками, как бы взбираясь по самому себе.
Также надо отметить, что при такой проблеме, как синдром Дюшенна, у ребенка постепенно атрофируются мышцы. Но нередко бывает так, что у крохи внешне мускулатура кажется очень развитой. Мальчик даже на первую вскидку оказывается как бы накачанным. Но это только лишь обман зрения. Все дело в том, что в процессе болезни мышечные волокна постепенно распадаются, а их место занимает жировая ткань. Отсюда и такой внушительный внешний вид.

Немного о деформации скелета
Если у ребенка прогрессирующая мышечная дистрофия Дюшенна, то постепенно у мальчика изменится форма скелета. Сначала патология затронет поясничный отдел, далее возникнет сколиоз, то есть произойдет искривление грудного отдела позвоночника. Позже проявится сутулость и, конечно же, изменится нормальная форма стопы. Вся данная симптоматика еще в большей степени будет сопутствовать ухудшению двигательной активности малыша.
О сердечной мышце
Обязательным симптомом при данной болезни также является поражение сердечной мышцы. Происходит нарушение ритма сердца, возникают регулярные перепады артериального давления. При этом сердце увеличивается в размерах. Но его функциональные возможности наоборот, уменьшаются. И в результате постепенно формируется сердечная недостаточность. Если эта проблема еще будет сочетаться с дыхательной недостаточностью, то возникает большая вероятность летального исхода.

Нарушения умственной активности
Нужно отметить, что мышечная дистрофия Дюшенна-Беккера не всегда проявляется таким симптомом, как умственная отсталость. Связано это может быть с дефицитом такого вещества, как аподистрофин, необходимого для работы головного мозга. Нарушения интеллекта могут быть самыми разными – от слабой умственной отсталости до идиотии. Усугублению этих когнитивных расстройств способствует еще и невозможность посещать садики, школы, кружки и иные места скопления детей. В результате возникает социальная дезадаптация.
Расстройства работы эндокринной системы
Различные эндокринные расстройства встречаются не более чем у 30-50% всех больных. Чаще всего это именно лишний вес, ожирение. При этом детки также имеют более низкий, чем у сверстников, рост.
Исход болезни
Какова клинико-эпидемиологическая характеристика мышечной дистрофии Дюшенна? Так, частота возникновения болезни – 3,3 пациента на 100 тысяч здоровых людей. Нужно отметить, что мышечная атрофия постепенно прогрессирует, и к 15-летнему возрасту мальчик уже не может обходиться без помощи окружающих, являясь полностью обездвиженным. Ко всему, происходит еще и частое присоединение различных бактериальных инфекций (чаще всего именно мочеполовой и дыхательной систем), при неправильном уходе за ребенком возникают пролежни. Если проблемы с дыхательной системой соединяются с сердечной недостаточностью, это грозит смертельным исходом. Если же говорить в общем, то такие пациенты практически никогда не живут более 30 лет.
Диагностика болезни
- Генетическое тестирование, то есть анализ ДНК.
- Электромиография, когда подтверждается первичное изменение мышц.
- Биопсия мышц, когда происходит определение наличия белка дистрофина в мышце.
- Анализ крови на определение уровня креатинкиназы. Нужно отметить, что именно этот фермент указывает на гибель мышечных волокон.
Лечение
Полностью излечиться от данной болезни невозможно. Можно только облегчить проявление симптомов, что сделает жизнь больного немного проще и удобнее. Так, после того, как пациенту ставят такой диагноз, чаще всего ему назначают терапию глюкокортикостеродами, которые призваны замедлить процесс развития болезни. Иные процедуры, которые также могут быть использованы при данной проблеме:
- Дополнительная вентиляция легких.
- Терапия медикаментами, которая направлена на нормализацию работы сердечной мышцы.
- Использование различных приспособлений, которые повышают мобильность пациента.
Также важно отметить, что сегодня ведутся разработки новейших методик, которые основаны на генной терапии, а также пересадке стволовых клеток.

Иные мышечные заболевания
Существуют еще и иные мышечные врожденные заболевания детей. К таким болезням можно отнести, помимо дистрофии Дюшенна:
- Дистрофию Беккера. Эта болезнь очень похожа на синдром Дюшенна.
- Мышечную дистрофию Дрейфуса. Это медленно прогрессирующая болезнь, при которой интеллект сохраняется.
- Прогрессирующую мышечную дистрофию Эрба-Рота. Проявляется в подростковом возрасте, прогрессирование быстрое, инвалидизация наступает рано.
- Плечелопаточно-лицевую форму Ландузи-Дежерина, когда мышечная слабость локализируется в области лица, плеч.
При этом надо отметить, что ни при одной из этих болезней не проявляется мышечная слабость у новорожденных. Вся симптоматика возникает в основном в подростковом возрасте. Длительность жизни пациентов чаще всего не превышает 30 лет.
24.1. Нервно-мышечные заболевания
Наследственные нервно-мышечные заболевания – большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата. Заболевания характеризуются мышечной слабостью, мышечными атрофиями, нарушениями статических и локомоторных функций.
При постановке диагноза учитываются возраст проявления первых клинических симптомов заболевания, локализация атрофии и характер распространения миодистрофического процесса (восходящий, нисходящий, наличие или отсутствие псевдогипертрофий, фасцикуляций, расстройств чувствительности, пароксизмов мышечной слабости), а также темп течения.
Прогрессирующие мышечные дистрофии – наиболее обширная группа. В зависимости от характера первичных изменений условно различают первичные (миопатии) и вторичные формы прогрессирующих мышечных дистрофий (денервационные амиотрофии – спинальные и невральные).
Наследственные пароксизмальные миоплегии – группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и плегиями. Наиболее распространенными из наследственных пароксизмальных миоплегии являются гипо-, гипер– и нормокалиемическая формы. Патогенез неясен. Предполагается генетически детерминированный дефект мембраны сарколеммы, нарушающий проницаемость для ионов натрия и калия,
Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля). Заболевание описано Вестфалем в 1895 г. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 6—15 лет. Пароксизмы характеризуются внезапным в ночные или утренние часы развитием мышечной слабости, обездвиженности, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами – лабильностью пульса, артериального давления, гипергидрозом. Приступы бывают парциальными, охватывающими небольшую группу мышц, и генерализованными. Во время приступа возникают нарушения сердечно-сосудистой деятельности: систолический шум, изменения ЭКГ. Сознание всегда сохранено. Средняя продолжительность приступа – несколько часов, крайне редко пароксизмы держатся несколько суток. Содержание калия в крови во время приступа менее 2 ммоль/л и ниже. Частота приступов вариабельна. Они провоцируются перееданием пищи, богатой углеводами, охлаждением, физическими нагрузками.
Лечение. Диета, богатая калием (чернослив, курага, картофель, изюм). Для купирования приступа назначают 10% раствор хлорида калия внутрь (по 1 столовой ложке каждый час) или 0,5% раствор в изотоническом растворе хлорида натрия внутривенно (2—2,5 г на 500 мл раствора в течение часа). Целесообразно применять также панангин внутривенно капельно.
Гиперкалиемическая форма пароксизмальной миоплегин (болезнь Гамсторп). Заболевание описано И.Гамсторп в 1956 г. Наследуется по аутссомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 1—5 лет. Симптоматика сходна с пароксизмами при гипокалиемической форме и характеризуется внезапным развитием мышечной слабости, плегиями, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами. В отличие от гипокалиемического гиперкалиемический паралич развивается обычно днем, сопровождается выраженными парестезиями, сочетается со слабостью мышц лица, артикуляционного аппарата, имеет меньшую продолжительность (30—40 мин). Во время приступа содержание калия в крови повышается до 6—7 ммоль/л. Частота приступов вариабельна: от ежедневных до нескольких раз в месяц. В межприступные периоды неврологическая симптоматика отсутствует. Провоцирующими факторами являются голодание, физические нагрузки, вызывающие утомление.
Лечение. Диета с повышенным содержанием углеводов, поваренной соли, ограниченным количеством калия. Вводят 40 мл 40% раствора глюкозы внутривенно вместе с инсулином подкожно; 20 мл 10% раствора хлорида кальция внутривенно.
Нормокалиемический (периодический) паралич. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется до 10-летнего возраста. Особенностью ее является сравнительно медленно (в течение нескольких суток) пароксизмально нарастающая умеренная слабость в мышцах туловища, конечностей и в жевательной мускулатуре, а также медленный (1—2 нед) регресс симптоматики. Провоцирующими факторами являются продолжительный сон, длительное пребывание в одной позе, переохлаждение.
Лечение. Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют. Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа, особенностей клинической картины, с учетом возраста, в котором начинается заболевание, времени возникновения пароксизма (ночью, утром, днем, в неопределенное время), степени выраженности мышечной слабости, частоты и длительности приступа, провоцирующих факторов, данных лабораторного биохимического исследования (содержание биоэлектрической активности мышц).
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний, – тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Лечение. Показана диета, богатая поваренной солью. Назначают диакарб.
24.2. Пирамидные и экстрапирамидные дегенерации
Хроническое прогрессирующее заболевание нервной системы, клинически проявляющееся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей.
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования при идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамин-?-гидроксилазы в сыворотке крови.
Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеобразного ядра, реже – в других базальных ганглиях.
Клинические проявления. Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично-генерализованные формы. В результате нарушения соотношения функции мышц-синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног. В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз.
Течение и прогноз. Заболевание в большинстве случаев неуклонно прогрессирует. Иногда отмечаются различной длительности ремиссии. Быстро происходит глубокая инвалидизация больных и наступает летальный исход, особенно при генерализованной форме.
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах.
Семейная атаксия Фридрейха – наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецессивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Патоморфология. Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени – Бурдаха, Флексига, Говерса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
Клинические проявления. Начало заболевания относится к 6—15-летнему возрасту. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически-мозжечковая. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен.
Заболевание медленно прогрессирует. Средняя продолжительность жизни 10—15 лет с момента его развития.
Диагностика и дифференциальный диагноз. Заболевание распознается на основании характерных симптомов – деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы и флексия концевых фаланг), поражения миокарда, эндокринных расстройств.
Дифференцировать заболевание следует от церебрального сифилиса, рассеянного склероза, фуникулярного миелоза и других форм мозжечковых дегенерации.
Лечение. Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Мозжечковая атаксия Пьера Мари – наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно-доминантный. Возникает заболевание в возрасте 20 лет и старше.
Патоморфология. Выявляется дегенеративное поражение клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста и продолговатого мозга.
Клинические проявления. Заболевание проявляется нарушениями функций мозжечка и его связей. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности (повышение сухожильных и периостальных рефлексов, клонусы стоп), а иногда с глазодвигательными нарушениями (косоглазие, птоз, недостаточность конвергенции). Характерным признаком является в различной степени выраженное снижение интеллекта.
Диагностика и дифференциальный диагноз. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии Фридрейха. Нужно учитывать тип наследования заболевания, возраст, в котором развиваются первые симптомы, характер изменения сухожильных рефлексов (при атаксии Фридрейха они снижены), наличие зрительных и глазодвигательных расстройств при атаксии Пьера Мари, деформации стоп и скелета. Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремитирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов.
Лечение. Симптоматическое.
Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов. По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенерации.
Тип I – оливопонтоцеребеллярная дегенерация Менделя.Наследуется по аутосомно-доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы.
Тип II – оливопонтоцеребеллярная дегенерация Фиклера—Винклера.Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается.
Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией.Наследуется по аутосомно-доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки.
Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана.Наследуется по аутосомно-доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно-суставного чувства и вибрационной чувствительности).
Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. Тип наследования аутосомно-доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами.
Дифференцировать оливопонтоцеребеллярные дегенерации следует от наследственной атаксии Фридрейха и Пьера Мари, прогрессирующих форм рассеянного склероза, опухолей мозжечка, ювенильных форм паркинсонизма.
Лечение. Симптоматическое. Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру.
Читайте также: