
Синдромы недостаточности костного мозга
Апластические анемии являются главным проявлением синдрома костно-мозговой недостаточности. Для такого состояния характерны:
- уменьшение объема гемопоэтической ткани;
- замещение костного мозга жировой тканью и фиброзной тканью (миелофиброз);
- панцитопения в периферической крови (выраженная анемия, Нb-20-30 г/л; нормохромия, макроцитоз, сниженное количество ретикулоцитов, повышенное содержание HbF, лейкопения, абсолютная нейтропения, относительный лимфоцитоз, тромбоцитопения, повышенная СОЭ);
- общеанемический синдром (бледность, вялость, одышка и др);
- иммунодефицитный синдром (инфекции, сепсис);
- геморрагический синдром (петехии, кровоподтеки, кровотечения);
- гемолитический синдром (короткоживущие эритроциты);
- увеличение содержания железа в сыворотке крови, как следствие нарушения включения железа в гемоглобин (насыщение им трансферрина достигает 100 %);
- высокий уровнь эритропоэтина в крови при сниженной эффективности его действия на костный мозг.
В таких случаях повреждаются клетки-предшественницы миелопоэза. Иногда формируются антитела к клеткам красного ряда, что дает основание предполагать аутоиммунный механизм развития такого рода анемий
Миелодиспластический синдром (МДС) — группа гетерогенных клональных заболеваний, характеризующаяся наличием цитопении в периферической крови, дисплазии в костном мозге и риском трансформации в острый лейкоз.
Этиология и патогенез:
Этиология миелодиспластических синдромов не установлена. Полагают, что генез их связан с изменениями гемопоэтической стволовой клетки и ее окружения. Вследствие этого выявляются морфологические дефекты клеток эритроцитарной, гранулоцитарной, лимфоцитарной и мегакариоцитарной линии гемопоэза, увеличение количества бластных клеток в периферической крови и костном мозге. Эти изменения встречаются с различной частотой и в разных сочетаниях. Наиболее постоянными являются дизгемопоэз, рефрактерная анемия, цитопения (1 -, 2-, 3-ростковая) и/или гипоплазия кроветворной ткани. Миелодиспластические синдромы обычно протекают в две фазы: гемодепрессии и клиники острого лейкоза (реже — хронического лейкоза или другой формы гемобластозов). Имеются сведения о кариологических, иммунологических нарушениях, изменениях антигенной структуры клеток, как правило, носящих мозаичный и неспецифический характер.
В основе патогенеза МДС лежит воздействие повреждающих факторов на полипотентную стволовую клетку, приводящее к появлению в ней генетических аномалий, а также феномена гиперметилирования ДНК. Указанные нарушения приводят к нарушению продукции клеток миелоидного ростка и появлению миелобластов в костном мозге и периферической крови, вследствие чего появляются диспластические изменения в зрелых клетках и их функциональная недостаточность, приводящие к описанным клиническим проявлениям. Феномен гиперклеточности костного мозга на фоне периферической цитопении объясняется ускоренным апоптозом аномально пролиферирующих клеток костного мозга.
Клиническая картина:
МДС отличает отсутствие типичной клинической картины. Симптоматику МДС составляют последствия дисмиелопоэза, то есть цитопении: анемия, нейтропения и тромбоцитопения (анемия Hb меньше 110 г/л, нейтрофилы меньше 1,800 на 1 микролитр крови; гематокрит меньше 36 % эритроцитов в общем объёме крови в организме; тромбоциты меньше 100,000 на 1 микролитр крови).
ФАБ-классификация:
Разработка этой системы классификации франко-американо-британской группой была начата в 1976 году и позже, в 1982 году, она приняла свой окончательный вид.
В основе классификации лежит ключевой для МДС синдром — рефрактерная, то есть устойчивая к лечению препаратами витамина В12 и фолиевой кислоты, анемия (РА). Четыре типа РА являются последовательными стадиями, с нарастанием тяжести МДС, что имеет своё отражение в прогнозе выживаемости. В этой связи появление в КМ бластов резко меняет прогноз выживаемости в худшую сторону.

Классификация ВОЗ:
В 2002 году Всемирная организация здравоохранения предложила новую классификацию миелодиспластических синдромов в 2008 году были сделаны предложения по её пересмотру.
Подгруппы, выделяемые в классификации ВОЗ включают: рефрактерную анемию и рефрактерную анемию с кольцевыми сидеробластами, рефрактерную цитопению с множественной дисплазией, рефрактерную анемию с избыточным количеством бластов-1 (содержание бластов в костном мозге составляет менее 10 %), рефрактерную анемию с избыточным количеством бластов-2 (содержание бластов в костном мозге превышает 10 %), синдром делеции 5q и миелодиспластический синдром неклассифицированный (с наличием или отсутствием кольцевых сидеробластов).
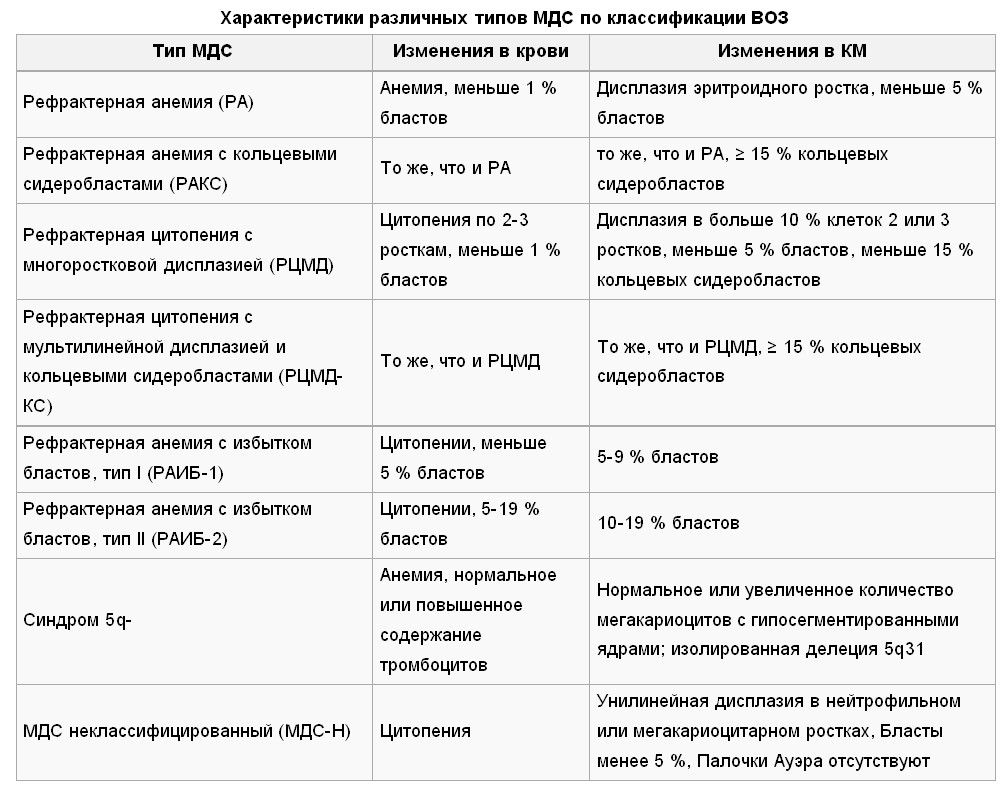
Минимальные диагностические критерии:
Минимальные диагностические критерии МДС включают обязательные диагностические условия — стабильная цитопения не менее 6 месяцев, (за исключением случаев, когда цитопения сопровождается специфическим кариотипом или дисплазией двух ростков кроветворения — в этих случаях длительность стабильной цитопении должна составлять не менее 2 месяцев), исключение других заболеваний, которые могут стать причиной развития дисплазии или/и цитопении.
В дополнение к этим двум диагностическим условиям для установления диагноза МДС необходимо соответствие хотя бы одному из трёх основных критериев:
дисплазия (≥ 10 % клеток одного или более из трёх основных ростков кроветворения в костном мозге).
содержание бластов в костном мозге 5-19 %.
специфический кариотип, например делеция (5q), делеция (20q), +8 или −7/делеция (7q).
Кроме того, для диагностики МДС используются дополнительные критерии, в том числе результаты проточной цитометрии, гистологического и иммуногистохимического исследования костного мозга, выявления молекулярных маркеров.
Морфологическое исследование биоптатов, полученных путём билатеральной трепанобиопсии, является полезным, помимо верификации диагноза самого МДС, с точки зрения дифференциальной диагностики с лимфопролиферативными и другими миелопролиферативными заболеваниями
При диагностике хромосомные аномалии обнаруживаются у 40-70 % пациентов с первичным MDS и у 95 % пациентов, MDS которых связан с терапией (вторичный).
К наиболее часто встречающимся при MDS цитогенетическим аномалиям относятся del(5q), −7 и +8
• Анемический;
• Геморрагический:
• Интоксикационный;
• Снижение резистентности к инфекциям.
Гиперластические синдромы
• Болезненность костей: трубчатых, позвоночника, суставов.
• Лимфаденопатия: увеличение любой группы лимфатических узлов, безболезненных при пальпации.
• Увеличение печени и селезёнки и образование экстрамедуллярных очагов кроветворения в них.
• Нейролейкемия. Поражение ЦНС возникает особенно часто при ОЛЛ и значительно ухудшает прогноз.
• Лейкемиды кожи (специфические узелки) чаще возникают при миеломонобластном и монобластном типах острого лейкоза.
Лейкозы подразделяют на острыеи хронические.Деление основано не на длительности течения (время начала болезни неизвестно), а в зависимости от степени зрелости лейкозных клеток. Присутствие в крови и костном мозге бластных клеток присуще острым лейкозам.
Название форм острого лейкоза происходит от названий нормальных предшественников опухолевых клеток: при остром лейкозе лимфобластный, миелобластный и т.д.; при хроническом - хронический миелолейкоз, хронический лимфолейкоз и т.д.
Точно идентифицировать бластные клетки по морфологическим признакам невозможно. Применяют ряд других, более надежных методов:
- гистохимические методы – выявление в клетках с помощью специальных красителей различных компонентов: гликогена, липидов, ферментов;
- цитогенетический метод – позволяет выявлять хромосомные аберрации;
• В-клеточные антигены: CD19, CD20, CD22;
• Т-клеточные антигены: CD2, CD3, CD4, CD8, CD5, CD7;
• Аг CD10 (CALLA, от англ. common acute lymphoid leukemia antigen,), присутствует на определенной стадии дифференцировки предшественников В-клеток.
- TДT (терминальная дезоксинуклеотидилтрансфераза) -фермент, присутствующий в лимфобластах, в миелобластах его нет.
• Основные морфологические признаки острых лейкозов:
Основные морфологические признаки хронических лейкозов:
- абсолютное преобладание клеток одного какого-то гемопоэтического ряда;
- отсутствие бластных клеток в периферической крови, небольшое их количество в костном мозге;
- при миелолейкозе в крови присутствуют все переходные формы дифференцировки гранулоцитов.
Острый миелобластный лейкоз
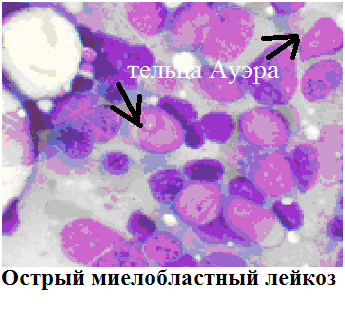
Острый миелобластный лейкоз (ОМЛ, острый нелимфобластный лейкоз) — самый частый вид острого лейкоза у взрослых. Средняя частота возникновения составляет 2,5:100 000 случаев в год. Симптомы острого миелоидного лейкоза типичны. Встречаются несколько разновидностей ОМЛ (см. классификацию), лечение и прогноз для них оказывается разным, уровень выживаемости на протяжении 5 лет 15-70% в зависимости от подвида.
Лечение химиопрепаратами до получения ремиссии, затем поддерживающее химиолечение или пересадка костного мозга.
М3 - острый промиелоцитарный лейкоз.

Без лечения это самый злокачественный вид лейкоза. Бластные клетки характеризуются обильной базофильной зернистостью, резко положительной реакцией на пероксидазу, цитогенетической аномалией t (15,17), секрецией тромбопластина, вследствие чего характерно развитие ДВС синдрома. В результате транслокации t (15,17) происходит слияние гена альфа рецептора ретиноевой кислоты (RARA) хромосомы 17 с PML геном хромосомы 15, образуется новый химерный ген. Этот ген кодирует синтез белка, который блокирует дифференцировку на стадии промиелоцита. Ретиноевая кислота, аналог витаминаА, снимает блок, вызывая дифференцировку промиелоцитов в зрелые нейтрофилы, которые имеют короткий срок жизни. Ретиноевую кислоту сочетают с химиотерапией антрациклином. Самый новый метод лечения – использование триоксида мышьяка.
Острый лимфобластный лейкоз (ОЛЛ) –
самое распространенное злокачественное заболевание у детей, заболеваемость составляет 1 на 50 000. Пик заболеваемости наблюдается и возрасте от 1 года до 6 лет. Редко ОЛЛ поражает также и взрослых.

Существуют два морфологических типа лимфобластов, однако в лечебном и прогностическом отношении морфологическая характеристика не существенна.
Фенотипические варианты ОЛЛ:
пре-В-клеточный тип (CD19, CD10), t(9,22) и TДT+, этот тип составляет 80% детских лейкозов;
пре-T-клеточный тип (CD5,CD7), TДT+;
В-клеточный тип (CD3).
Клиническая характеристика ОЛЛ у детей
Интоксикационный синдром проявляется астенией, лихорадкой, потерей массы тела у детей раннего возраста. Дисфункция костного мозга проявляется всеми типичными синдромами. Инфильтрация печени и селезенки может проявляться болями в животе, тошнотой, анорексией. Лейкемическая инфильтрация, инфаркты костей вызывают боли в костях, переломы трубчатых костей или позвоночника. Увеличение яичек у мальчиков отмечается в 5–30% случаев ОЛЛ. Центральная нервная система обычно вовлекается при рецидивах заболевания после химиотерапии.
Лечение выстраивается в зависимости от возраста, иммунофенотипа бластных клеток, раннего ответа на терапию, генотипических характеристик лейкемических клеток и кинетики исчезновения остаточной опухолевой популяции.
Хронический лимфолейкоз -второй по частоте лейкоз,встречается в пожилом возрасте, у мужчин в два раза чаще. Содержание лимфоцитов в крови доходит до 80% - 99%, увеличиваются лимфатические узлы, печень и селезенка. Иногда масса селезенки составляет несколько килограммов.

Заболевание протекает длительно с высокими показателями выживаемости. Клетки аномального клона являются зрелыми лимфоцитами, в основном В-лимфоцитами (приблизительно в 95% случаев), реже Т-лимфоцитами. Функциональная неполноценность лимфоцитов приводит к возникновению у больных инфекционных и аутоиммунных осложнений (анемия, тромбоцитемия). Т-клеточные формы имеют более агрессивное течение.
Характерно присутствие в мазках крови разрушенных лимфоцитов - так называемых клеточных теней (тени Боткина-Гумпрехта).

На ранних стадиях лечение не проводится. Многие пациенты ведут нормальную и активную жизнь годами.
Лечение начинают в той стадии, когда заболевание может повлиять на качество жизни пациента. Применяется химиотерапия, радиотерапия, иммунотерапия, трансплантация костного мозга. В настоящее время лейкоз считается неизлечимым.
Существует группа редких опухолей, основным проявлением которых служит лимфоцитоз. К ним относится и волосатоклеточный лейкоз - B-клеточное новообразованиенизкой степени злокачественности. Клетки покрыты ворсинками, похожими на волосы. 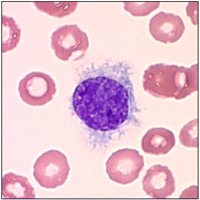
Клинические проявления варьируют от бессимптомного лимфоцитоза и спленомегалии до тяжелых инфекций, обусловленных резкой нейтропенией.
Аналоги пуринов, особенно кладрибин, совершили переворот в лечении этого заболевания. Частота полных ремиссий при лечении кладрибином превышает 80%, полная ремиссия нередко достигается уже после первого курса и часто длится более 3 лет.
Хронический миелолейкоз– лейкозный клон возникает из полипотентной стволовой клетки, поэтому в патологический процесс вовлечены клеточные элементы всех рядов гемопоэза. Ph-хромосома обнаруживается почти во всех делящихся клетках миелопоэза, а также в бластных клетках при лимфобластных кризах.
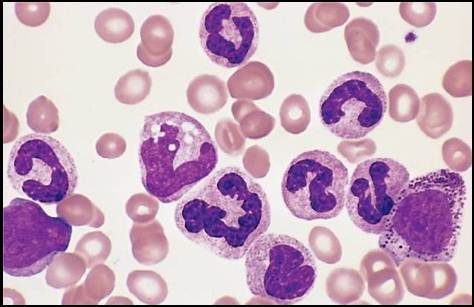
Пролиферирует в основном гранулоцитарный росток. Количество лейкоцитов в периферической крови от 20 до 500×10 9 /л с гиперрегенеративным сдвигом влево. Если количество незрелых форм невелико, необходимо проводить дифференциальную диагностику с лейкемоидной реакцией миелоидного типа.
Дифференциальные признаки миелолейкоза: наличие Ph-хромосомы; увеличение содержания в крови базофилов и эозинофилов (базофильно-эозинофильная ассоциация), резкое снижение в лейкоцитах щелочной фосфатазы.
Лейкоз медленно прогрессирует, моноклоновая опухоль превращается в поликлоновую. В финальной фазе развивается так называемый бластный криз.
Лечение. Транслокация t(9;22) формирует ген bcr-abl, продуктом которого является тирозинкиназа. В конце 1990 годов начали применение ингибитора тирозинкиназ STI-571(imatinib, Gleevec). Он тормозит пролиферацию аномальных клеток. Ныне получены более мощные ингибиторы тирозинкиназ dasatinib и nilotinib, их применение кардинально изменило судьбу больных.
В клиническом плане хронический лимфолейкоз, промиелоцитарный лейкоз и волосатоклеточный лейкоз принято рассматривать как отдельные морфологические и клинико-патологические единицы, требующие различных терапевтических подходов.
Принципиальная возможность излечения большинствадетей, больных острым лимфобластным лейкозом, - одно из наиболее ярких достижений последних десятилетий. Современное эффективное лечение хронического миелолейкоза, острого промиелоцитарного лейкоза требует пожизненного регулярного приема лекарств. Оно позволяет не только спасти людям жизнь, но и сохраняет их трудоспособность на продолжительное время.
Лейкемоидные реакции - патологические изменения состава крови, сходные с картиной крови при лейкозах. Вызывать лейкемоидные реакции могут вирусы, токсины тканевых гельминтов, продукты распада клеток крови (при гемолизе) и опухолей, сепсис и др. При этом происходит гиперплазия кроветворных клеток при нормальных соотношениях отдельных элементов в красном костном мозге.
Лейкемоидные реакции могут быть миелоидного, эозинофильного, лимфоидного, моноцитарного типа, к ним также относятся симптоматические эритроцитозы.
Лейкемоидные реакции миелоидного типанапоминают хронический миелолейкоз. Это наиболее частый тип лейкемоидных реакций. Причинами могут быть инфекции, шок, ионизирующее излучение, интоксикации (приём сульфаниламидных препаратов, лечение глюкокортикоидами, уремия, отравление угарным газом). В периферической крови выявляют умеренный лейкоцитоз с гиперрегенераторным сдвигом нейтрофильного ядра влево, с токсической зернистостью и дегенеративными изменениями нейтрофильных гранулоцитов. Миелограмма характеризуется увеличением содержания молодых клеток нейтрофильного ряда, с преобладанием более зрелых элементов (миелоцитов, метамиелоцитов). Активность щелочной фосфатазы в нейтрофилах повышена.
Лейкемоидные реакции эозинофильного типа.Причинами возникновения этого типа реакций служат в основном гельминтозы, реже коллагенозы, лимфогранулематоз, эндокринопатии. Характерен лейкоцитоз до 40-50×10 9 /л, эозинофилия (60-90%) за счёт зрелых форм эозинофилов. Исследование костного мозга позволяет дифференцировать этот тип реакции с эозинофильным вариантом хронического миелолейкоза.
Лейкемоидные реакции лимфоидного и моноцитарного типанаблюдаются при инфекционном мононуклеозе- вирусном заболевании, проявляющемся изменениями крови, реактивным лимфаденитом и увеличением селезёнки. В периферической крови наблюдают лейкоцитоз до 10-30×10 9 /л. Содержание лимфоцитов достигает 50-70%, моноцитов - 10-40%, появляются плазматические клетки, атипичные мононуклеары, патогномоничные для данного заболевания.
ПАТОЛОГИЯ СИСТЕМЫ ГЕМОСТАЗА
Система гемостаза выполняет в организме две важные функции: 1. собственно гемостаз – остановку кровотечения при повреждении сосуда; 2. поддержание жидкого состояния крови. Первую функцию называют гемостатической, вторую – антигемостатической.
Гемостаз достигается взаимодействием между стенкой сосуда (сосудистый компонент), тромбоцитами (тромбоцитарный или клеточный компонент) и белками крови, входящими в состав свертывающей, противосвертывающей и фибринолитической систем (плазменный или коагуляционный компонент).
Сосудистое звено (компонент) гемостаза.Спазм сосуда— самая ранняя реакция на повреждение, сначала рефлекторная, затем усиленная и продленная благодаря секреции тромбоцитами активных вазоконстрикторов: тромбоксанаА2 (ТХА2), серотонина и эндотелина-1, вырабатываемого эндотелием. Все компоненты сосудистой стенки участвуют в реакции на травму, особенно важную роль в гемостазе играет эндотелий. Эндотелиальные клетки вырабатывают компоненты базальной мембраны; коллаген; белки, необходимые для прилипания тромбоцитов к коллагену, вещества, которые секретируются в кровь и регулируют текучесть крови. Кроме того, они продуцируют вещества, расширяющие сосуды и препятствующие адгезии и агрегации тромбоцитов: простациклин (ПГ I2) и монооксид азота (NO). В месте повреждения эндотелиоциты вырабатывают сосудосуживающий фактор эдотелин-1, а продукция ими простагландина ПГ I2 и NO уменьшается. Фактор Виллебранда (ФВ) синтезируется практически только в эндотелиоцитах, его продукция возрастает при повреждении эндотелия, вследствие чего этот фактор используется в качестве маркера повреждения эндотелия.
Вне повреждения эндотелий является антигемостатической, антитромботической поверхностью, а после травмы превращается в гемостатическую поверхность, фиксирующую тромбоциты и факторы свертывания, секретирующую в кровь множество активных веществ.
Клеточное звено системы гемостаза. Тромбоциты образуются в костном мозге в результате фрагментации мегакариоцитов. Они имеют форму дисков диаметром 2-5 мкм. Их число в крови здорового человека составляет 180 – 320 х 10 9 /л.
Тромбоцитам принадлежит ведущая роль в первичной остановке кровотечений из микрососудов. Тромбоциты циркулируют в кровотоке, не взаимодействуя друг с другом и с эндотелием сосудов. При нарушении целостности сосуда они накапливаются в местах повреждения и формируют так называемый белый или тромбоцитарный тромб. Такой тромб впоследствии закрепляется нитями фибрина, которые образуются в результате активации свертывающей системы. Так формируется полноценная гемостатическая пробка и происходит остановка кровотечения.
Стадия адгезии - при повреждении сосудистой стенки обнажаются компоненты, расположенные под эндотелием, способные вызвать прилипание тромбоцитов, их активацию. На волокнах коллагена фиксируются молекулы фактора Виллебранда (ФВ) и к ним, в свою очередь, прикрепляются тромбоциты. Процессы прикрепления клеток друг к другу называют термином "клеточная адгезия". Она обеспечивается специальными клеточными рецепторами, мембранными гликопротеидами, получившими название молекул клеточной адгезии. Гликопротеиды (ГП) тромбоцитов обеспечивают протекание начальных стадий процесса тромбообразования. Это так называемый первичный гемостаз — обратимый процесс, на который не распространяется действие гепарина. В начальной стадии адгезии тромбоцитарный гликопротеид ГП 1b взаимодействует с фактором Виллебранда, фиксированным на коллагене, а гликопротеид ГП 1a – непосредственно с коллагеном. Позднее процесс адгезии усиливает гликопротеидный комплекс, появляющийся на мембранах тромбоцитов в результате их активации - ГП IIb-IIIa.
Взаимодействие тромбоцитов с коллагеном, ФВ и тромбином приводит к их активации.
В результате активации происходит ряд важных процессов: секреция множества активных веществ; изменение формы тромбоцита; транспорт на мембрану тромбоцитов фосфолипидов, обеспечивающих каталитическую поверхность для активированных IX и Х факторов свертывания; транспорт на поверхность тромбоцитов Р-селектина, обеспечивающего фиксацию лейкоцитов в зоне повреждения; сокращение (ретракция) тромба.
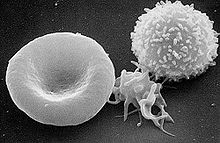
Сканирующая электронограмма. Слева направо: эритроцит, активированный тромбоцит, лейкоцит.
Самое раннее из этих событий – дегрануляция и секреция АДФ. Кроме того, из липидного слоя мембраны освобождается арахидоновая кислота, которая под влиянием фермента циклооксигеназы превращается в простагландины, затем под влиянием тромбоксансинтетазы – в тромбоксан А2 (ТхА2). ТхА2 – мощный стимулятор тромбоцитов, вовлекает новые порции тромбоцитов в процесс агрегации. Ингибиторы циклооксигеназы, такие как аспирин, блокируя синтез ТхА2, ингибируют агрегацию тромбоцитов. ТхА2 , АДФ и тромбин (он чуть позднее появляется в процессе свертывания крови) – агреганты. Агреганты изменяют конформацию гликопротеидного комплекса ГП IIb-IIIa, он становится рецептором фибриногена, обеспечивая склеивание тромбоцитов.
Агрегация тромбоцитов. Ключевая роль в процессе агрегации тромбоцитов принадлежит комплексу гликопротеидов IIb-IIIa. Он приобретает способность к связыванию фибриногена только при активации тромбоцитов. Фибриноген, взаимодействуя одновременно с двумя молекулами гликопротеида IIb-IIIa, образует молекулярные "мостики" между тромбоцитами, обеспечивая их склеивание. Сначала связь между тромбоцитами непрочная – это обратимая агрегация, затем изменение формы тромбоцитов, состояния их цитоплазмы обеспечивают необратимую агрегацию.
Активированные тромбоциты на протяжении всего процесса тромбообразования секретируют содержимое гранул через систему открытых каналов - стадия секреции. Тромбоциты имеют три типа гранул: α-гранулы, плотные гранулы и лизосомы. Плотные гранулы содержат АДФ, АТФ, серотонин, пирофосфат, ионы Са 2+ ; α-гранулы – тромбоцитарный фактор роста, трансформирующий фактор роста β1, ß-тромбоглобулин, фактор VIII, антиген фактора Виллебранда, фактор V, фибриноген, тромбоспондин, фибронектин; лизосомальные гранулы - фосфатазы, арилсульфатазы, кислые гидролазы.
Образование тромба – лишь временное решение проблемы остановки кровотечения, необходимо восстановление поврежденного сосуда. Тромбоциты помогают этому, секретируя факторы роста, стимулирующие рост сосудов; хемоаттрактанты, привлекающие к месту повреждения фибробласты из окружающей ткани.
Последнее изменение этой страницы: 2016-08-15; Нарушение авторского права страницы
Наследственные синдромы недостаточности костного мозга почти всегда обнаруживаются в детском возрасте симптомами, связанными с цитопенией, или характерными физическими аномалиями. Недостаточность костного мозга, часто скрытая в начальном периоде, к моменту распознавания может проявляться как угнетение одного или нескольких ростков кроветворения.
Анемия Фанкони. Классический фенотип анемии Фанкони имеет следующие признаки: семейная апластическая анемия (аутосомно-рецессивный тип наследования), низкорослость, отсутствие либо гипоплазия большого пальца или лучевой кости, микроцефалия и гипопигментированные пятна (“кофе с молоком”).
Гематологические проявления при анемии Фанкони могут варьироваться от нормального количества клеток крови с небольшим повышением уровня гемоглобина Р до тяжелой панцитопении с полной зависимостью больного от гемотрансфузий. Аналогично в костном мозге может наблюдаться снижение клеточности от умеренной до сильной степени. Первая линия поддерживающей терапии — андрогены, которые эффективны приблизительно у 50 % пациентов. Вначале обычно реагирует эритроидный росток (в течение 1-2 мес), затем гранулоцитарный; последним повышается содержание тромбоцитов, что наименее надежно и может наблюдаться только через год. У большинства пациентов после прекращения введения андрогенов возникают рецидивы. Трансплантация костного мозга от подходящего совместимого донора является в настоящее время единственным радикальным методом терапйи, при котором удается добиться 5-летнего выживания 2/з пациентов. Обычная высокодозная химиотерапия, назначаемая перед трансплантацией костного мозга, должна быть ограничена из-за чувствительности к ней гемопоэтических клеток при анемии Фанкони (глава 8). В будущем может оказаться полезной генная терапия аутологичного костного мозга.
Пациенты без цитопений имеют значительный риск развития новообразований, включая лейкоз (обычно миелоидный), предлейкоз и карциному (рак) (в частности, сквамозный и гепатоцеллюлярный). Общая частота возникновения злокачественных новообразований приближается к 20 %. После трансплантации костного мозга часто образуются эпителиальные опухоли, хотя они не всегда угрожают жизни пациента.
Анемия Даймонда-Блэкфана. Изолированную врожденную недостаточность эритропоэза называют анемией Даймонда-Блэкфана (АДБ). Диагностическими критериями являются: 1) ретикулоцитоления с макроцитарной или нормо- цитарной анемией; 2) нормальная клеточность костного мозга с изолированной эритроидной гипоплазией и 3) нормальное или близкое к нормальному количество лейкоцитов и тромбоцитов.
Транзиторная детская эритробластопения. В соответствии с названием транзи- торная детская эритробластопения (ТДЭ) подразумевает резкое, но временное подавление эритропоэза. Такое угнетение — отличительный признак ТДЭ, однако окончательный диагноз часто ставится ретроспективно. Вероятной причиной заболевания является вирусная инфекция клеток эритроидного ростка, хотя тест на обнаружение парвовируса редко бывает положительным. Проявления синдрома включают изолированную анемию, ретикулоцитопению и обнаруживаемую при исследовании костного мозга глубокую гипоплазию эритроидного ростка, т. е. аналогичны таковым при АДБ. Как и при АДБ, миелоидный и мегакариоци- тарный ростки существенно не повреждены. Различия между двумя синдромами устанавливают на основании клинических данных. Первые признаки проявления ТДЭ обычно наблюдаются у детей старше 2 лет. Наличие в анамнезе вирусных инфекций подтверждает диагноз ТДЭ (хотя, конечно, у детей с АДБ также развиваются вирусные инфекции, иногда непосредственно перед появлением симптомов). В течение острой, ретикулоцитопенической, фазы ТДЭ маркеры напряжения эритропоэза (стрессорного эритропоэза) вообще отсутствуют, что является диагностическим критерием; иными словами, обнаружение данных маркеров в этой фазе свидетельствует о развитии АДБ. К маркерам напряжения эритроцита относятся увеличение среднего объема эритроцитов, уровня гемоглобина Р и уровня антигена I. Однако эти маркеры могут выявляться в течение восстановительного периода, что затрудняет дифференциальный диагноз ТДЭ и АДБ; но нормализация количества ретикулоцитов у пациента, не получавшего стероидов, позволяет исключить диагноз АДБ. Таким образом, если лечащий врач имеет серьезные основания предполагать у пациента ТДЭ, то больного можно просто наблюдать. При постановке этого диагноза восстановление произойдет в течение 1-2 месяцев. Пациенты часто удовлетворительно переносят снижение уровня гемоглобина до 50 г/л, не нуждаясь в трансфузионной поддержке.
Тромбоцитопения с отсутствием лучевых костей. Характерная физическая аномалия при этом редко встречающемся синдроме — отсутствие лучевых костей (двустороннее) при наличии больших пальцев (при анемии Фанкони большие пальцы отсутствуют). Заболевание обнаруживается у младенцев в первые недели жизни на основании выраженной физической аномалии или из-за геморрагических проявлений. Характер наследования аутосомно-рецессивный. При исследовании костного мозга, наряду с нормальным миело- и эритропоэзом, регистрируется отсутствие, гипоплазия или незрелость развивающихся,мегакариоцитов. Анемия с ретикулоцитозом может возникнуть вследствие кровавой диареи, связанной с потреблением коровьего молока.
Нередко наблюдается лейкемоидная реакция с резко увеличенным количеством лейкоцитов. Эффективны лишь переливания тромбоцитов. Если пациенты переживают первый год, что происходит в большинстве случаев, долгосрочный прогноз положителен, при этом количество тромбоцитов обычно стабильно превышает 100 000/мкл.
Амегакариоцитарная тромбоцитопения. Амегакариоцитарная тромбоцитопения — гипомегакариоцитарный аналог анемии Даймонда-Блэкфана. Лучевые кости — в наличии; физические аномалии — лишь в качестве исключения. Тромбоцитопения у младенца в основном проявляется в течение первых нескольких месяцев жизни, при этом в костном мозге наблюдается селективное угнетение мегакариоцитов. Характер наследования рецессивный, в некоторых случаях аутосомный, в других — сцепленный с Х-хромосомой. Благотворное влияние на течение заболевания оказывают кортикостероиды и/или андрогены, хотя средний срок выживания составляет только 5 лет. У многих пациентов развивается апластическая анемия. Эффективна трансплантация костного мозга.
Мужчина в возрасте 28 лет, почувствовав слабость, пришел на прием к терапевту. За несколько недель до появления этого ощущения у пациента отмечались головные боли, недомогание, миалгия, лихорадка и озноб. За два-три дня до визита к врачу появилась диффузная яркая макуло-папу- лезная сыпь и боли в суставах. Уже около 1 недели пациент ощущает слабость и утомляемость, а в последние несколько дней — постоянную одышку при обычной физической нагрузке, без лихорадки, потливости или озноба. При физикальном обследовании выявлена бледность, не связанная с каким-либо острым состоянием. Частота дыхания составляет 22/мин, пульс регулярный с частотой 100/мин. Температура тела нормальная. Выраженная бледность кожи и слизистых. Сыпи нет, лимфаде- нопатия не выявлена. Сердце и легкие без патологии. Живот мягкий. Конечности не изменены. При ректальном обследовании — стул коричневый, без скрытой крови. Нервная система без изменений. В моче отсутствуют эритроциты и билирубин. Данные клинического анализа крови: гемоглобин — 50 г/л, гематокритное число —15 % при среднем объеме эритроцита 93 мкм3; лейкоциты — 4500/мкл, лейкоцитарная формула в норме; количество тромбоцитов — 320 000/мкл. В мазке периферической крови внешний вид тромбоцитов и лейкоцитов нормальный. Окраска и форма эритроцитов — без отклонения от нормы. Полихромазия не выявлена.
Вопрос 1. Каков следующий этап обследования?
Ответ. Детально расспросить пациента с целью выявить признаки возможного кровотечения из желудочно-кишечного, мочеполового тракта или слизистых. Выяснить, была ли у пациента темная моча; это позволит определить, имелись ли в прошлом гемолитические эпизоды с бийирубинурией.
В данном случае никакой новой информации получено не было.
Вопрос 2. Что выполнено далее?
Ответ. Определено количество ретикулоцитов, а также активность ЛДГ, содержание гаптоглобина, билирубина в крови и гемосидерина в моче. Полное отсутствие ретикулоцитов. Другие исследования не выявили никаких аномалий.
Вопрос 3. Как интерпретировать результаты обследования?
Ответ. Костный мозг не реагирует адекватно на анемию, так как нет продукции молодых клеток (ретикулоцитов). Признаков гемолиза при лабораторном обследовании не обнаружено. Нормальные показатели активности ЛДГ и содержания билирубина и гаптоглобина указывают на то, что в последнее время не было каких-либо гемолитических эпизодов. Нормальный уровень гемосидерина в моче свидетельствует об отсутствии в ближайшем анамнезе больного гемолиза (в противном случае гемосидерин откладывался бы в клетках почечных канальцев, которые при слущивании привели бы к появлению в моче железосодержащих пигментов).
Вопрос 4. Каков следующий шаг?
Ответ. Выполнено исследование костного мозга. Цитоз оказался нормальным. Предшественники тромбоцитов и лейкоцитов в границах нормы. Однако выявлено очень мало всех видов предшественников эритроцитов. Обнаружено несколько гигантских пронормобластов, которые содержали внутриядерные включения свободного аморфного розового материала. Положительными оказались дополнительный анализ крови на наличие генома парвовируса В19 (анализ ДНК методом дот-блот-гибридизации) и тест на ВИЧ.
Вопрос 5. Каков вероятный диагноз?
Ответ. Больной, скорее всего, инфицирован парвовирусом В19 на фоне ВИЧ-инфекции.
Вопрос 6. Как лечить анемию у этого пациента?
Ответ. Пациенту было проведено переливание эритроцитов. Вводился также внутривенный иммуноглобулин.
У пациента были определены количество СБ4+-клеток и вирусная нагрузка. Начато соответствующее лечение ВИЧ-инфекции. В течение следующих 2-х недель развился ретикулоцитоз и временно повысилось гематок- ритное число. Однако для сохранения его на приемлемом уровне было назначено постоянное введение иммуноглобулина.
Акег В. Р. (ес!.). РеппаЫ НетаЫо&у. Ые\уУогк: СЬигсЬШ Ьтп^опе, 1989.
Эта монография включает исчерпывающие сведения по эпидемиологии, генетике, истории, а также обзор клинических наблюдений, методов лабораторной диагностики и лечения синдромов недостаточности костного мозга, в основном конституциональных, которые развиваются на ранних этапах жизни.
НоЯшап К. Н. еЬ а1. (ес!з). Нета1о1о%у: Вазгс Рппсгр1ез апй Ргасйсе, 2пс1 ес1. Уогк: СЬигсЬШ Ьтп^зите, 1995.
Фундаментальный учебник по гематологии, в котором обсуждаются фактически все синдромы недостаточности костного мозга, представленные в данной главе. Приводятся полезные диагностические и терапевтические схемы, хотя они не являются единственно возможными.
ЗсЬгоейег-КигСЬ Т. М., АиегЬасЬ А. Б., ОЪе С. (ейз). Рапсопг Апетга: СНпгса1', Су1о&е- пеЫс, апй ЕхрептепШ А$рес1$. Уогк: 5рпп§ег-Уег1а§; 1989.
В книге представлен полный спектр клинических вариантов анемии Фанкони (АФ) с рассмотрением анатомических аномалий, примеров наследования, возраста манифестации заболевания, вариантов лечения. Однако в ней отсутствуют последние данные о молекулярных основах АФ и применении смягченных режимов трансплантации.
Уоип§ N. 5., Акег В. Р. Ар1азЫс Апетга, Асдиггей апй ЫкепЬей. РЫ1ас1е1рЫа: \\Л В. 5аип- Йегз; 1994.
Это полное руководство, в котором много сведений о патофизиологии и молекулярных механизмах синдромов недостаточности костного мозга, а также рассмотрена возможность использования большого количества современных терапевтических средств, таких как факторы роста, при лечении АА.
Уоип§ N.. Ке15и М. (ейз). Бги^ геЫ;ес1 Ыоос!
Читайте также: