
Системной красной волчанке склеродермии и ревматоидному артриту
Рассмотрены вопросы поражения легких при системной красной волчанке, ревматоидном артрите, системной склеродермии, полимиозите/дерматомиозите, синдроме Шегрена, смешанном заболевании соединительной ткани, гранулематозе Вегенера, синдроме Черджа–Стросса, с
Aspects of lungs lesion with exanthematous lupus erythematosus, rheumatoid arthritis, Addison’s keloid, polymyositis/dermatomyositis, Sj?gren’s sicca syndrome, mixed connective tissue disease, necrotizing respiratory granulomatosis, Churg–Strauss syndrome, Goodpasture’s syndrome and Bechterew’s disease have been analysed.
В основе поражения легких (ПЛ) при системных аутоиммунных заболеваниях (САЗ) лежат расстройства механизмов иммунорегуляции и гиперреактивность организма. Макрофаги и лимфоциты являются ключевыми клетками, участвующими в инициации и сохранении иммунного ответа в легких. Альвеолярные макрофаги — резиденты легочной ткани — поглощают чужеродные агенты, проникшие через слизистые поверхности легких и бронхов. Кроме того, эти клетки служат в качестве антигенпредставляющих клеток для Т-лимфоцитов. Относительно небольшое число лимфоцитов присутствуют в нормальной паренхиме легких. Однако после стимуляции соответствующим антигеном окружающей лимфоидной ткани лимфоциты мигрируют в легкие и принимают участие в воспалительной реакции.
Проведение специальных исследований, включавших компьютерную томографию высокого разрешения (КТВР) и биопсию легкого, выявило, что при САЗ встречаются различные гистологические формы поражения интерстиция легких (табл.).
Системная красная волчанка (СКВ) — системное аутоиммунное заболевание неизвестной этиологии, характеризующееся гиперпродукцией органоспецифических аутоантител к различным компонентам клеточного ядра с развитием иммуновоспалительного повреждения тканей и внутренних органов.
Ревматоидный артрит (РА) — системное аутоиммунное заболевание соединительной ткани с преимущественным поражением суставов по типу хронического прогрессирующего эрозивно-деструктивного полиартрита и внесуставными проявлениями.
ПЛ при РА было впервые описано в 1948 г., когда Эллман и Болл установили диффузный фиброз легких у трех пациентов. Легочные проявления при РА включают поражение плевры, образование ревматоидных узелков в легких, ИПЛ, легочный васкулит, альвеолярные кровоизлияния, бронхообструктивные изменения. Почти у половины больных РА при вскрытии выявляется плеврит. Экссудативный плеврит может быть односторонним или двусторонним и сохраняться в течение многих месяцев. Наличие плеврального выпота вызывает нарушение легочной функции. Как правило, плевральная жидкость бывает экссудативной с низким содержанием глюкозы и низким уровнем комплемента. Чаще у пациентов с РА встречается сухой плеврит и является находкой при рентгенологическом исследовании.
Ревматоидные узелки (РУ), как единичные, так и множественные, могут быть обнаружены в легочной паренхиме больных РА. РУ легких у пациентов с РА были впервые описаны Каплан в 1953 г. РУ могут появиться до, во время или после начала РА. ИПЛ характеризуется хроническим воспалением стенок альвеол и скоплением больших мононуклеарных клеток в альвеолах. Довольно часто у больных РА встречается сочетание ИПЛ и подкожных ревматоидных узелков. Прогноз для пациентов с РА с ИПЛ неблагоприятен. Легочный васкулит является одним из редких легочных проявлений РА. Альвеолярные кровоизлияния, не часто встречающиеся у больных РА, приводят к кровохарканью и анемии. У пациентов с РА отмечается высокая частота поражения бронхов. ПЛ может быть результатом токсического воздействия препаратов, используемых в терапии РА. Для того чтобы обеспечить оптимальный эффект от лечения, врач всегда должен учитывать возможную легочную патологию при оценке состояния пациентов с РА [1–3].
Системная склеродермия (ССД) — диффузное заболевание соединительной ткани с прогрессирующим фиброзом, распространенными вазоспастическими нарушениями и характерными изменениями кожи, опорно-двигательного аппарата и внутренних органов. Среди висцеральных проявлений ССД ПЛ занимает важное место.
Плеврит при ССД наблюдается реже, чем при РА и СКВ. В то же время у больных ССД значительно выше частота ИПЛ, при этом морфологическое исследование показывает выраженный фиброз интерстициальной ткани и утолщение межальвеолярных перегородок. Исследование функции внешнего дыхания (ФВД) демонстрирует снижение диффузионной способности легких (ДСЛ) даже при отсутствии каких-либо клинических симптомов и рентгенологических изменений.
У пациентов с ССД выявляется рестриктивный тип нарушения вентиляции легких. У большинства больных ССД с ЛФ наблюдается гистологическая картина обычной интерстициальной пневмонии (ОИП), однако во многих случаях встречается гистопатологический тип неспецифической интерстициальной пневмонии (НИП).
Развитие ЛФ инициируется микрососудистыми изменениями, которые приводят к повреждению эндотелиальных клеток и поражению альвеолярного эпителия. Это приводит к активации каскада свертывания (рис.).
Экспрессия аутоантител является предиктором поражения внутренних органов, в частности ПЛ. Наличие антител к Scl-70 (антисклеродермальные антитела с молекулярной массой 70 кДа, антитела к топоизомеразе I) в значительной степени является фактором риска развития ИПЛ, в то время как антицентромерные антитела (АЦА) говорят о низкой вероятности развития рентгенологических признаков ЛФ.
У пациентов с ССД встречается легочная гипертензия (ЛГ). ЛГ может быть изолированной, возникшей вследствие поражения сосудов, или вторичной — при поражении паренхимы легких или левых отделов сердца. ЛГ развивается у 5–7% больных, чаще на поздних стадиях лимитированной формы ССД. Предиктор ЛГ — изолированное уменьшение ДСЛ. Более редко встречаются аспирационная пневмония, причиной которой является дисфункция пищевода, а также спонтанный пневмоторакс, лекарственный пневмонит [1–3, 6–8].
Полимиозит/дерматомиозит (ПМ/ДМ) — группа хронических диффузных заболеваний поперечнополосатой мускулатуры, основным проявлением которых выступает мышечная слабость.
Около 40% пациентов с ПМ/ДМ имеют патологию легочной системы. Одной из причин этого является поражение мышц. Поражение межреберных мышц и высокое стояние диафрагмы приводят к уменьшению экскурсии грудной клетки и появлению вентиляционных нарушений по рестриктивному типу. В отличие от других САЗ, ПЛ при ПМ/ДМ не затрагивает в первую очередь дыхательные пути или плевру. Наиболее распространенной патологией является аспирационная пневмония, которая возникает по причине слабости мышц глотки и верхней трети пищевода.
Болезнь Шегрена (БШ) — системное аутоиммунное заболевание неизвестной этиологии, характеризуется поражением секретирующих эпителиальных желез, с вовлечением преимущественно слюнных и слезных желез (ксеростомия, ксерофтальмия).
Поражение экзокринных желез верхних дыхательных путей часто приводит к сухости носовых ходов и бронхов. Наиболее распространенным проявлением ПЛ, связанным с БШ, является лимфоцитарный пневмонит, поражающий нижние доли. У пациентов с БШ может развиться плеврит (с или без выпота), ИПЛ с очагами лимфоидной инфильтрации. При развитии неходжкинских лимфом метастатические поражения легких встречаются часто, реже наблюдают формирование MALT-ткани (mucosal-associated lymphoid tissue) с развитием первичной MALT-лимфомы [1–3, 12, 13].
Смешанное заболевание соединительной ткани (синдром Шарпа) (CЗСТ) — аутоиммунное заболевание, характеризующееся наличием отдельных признаков СКВ, ССД, РА, ПМ/ДМ в сочетании с высоким титром антител к экстрагируемому ядерному антигену — U1-RNP.
ИПЛ и ЛГ довольно часто встречаются у больных с СЗСТ, при этом нередко имеют субклиническое течение. Обследование пациентов с СЗСТ показало повышение уровня иммунных комплексов (ИК) и увеличение потребления комплемента. ИК-опосредованное повреждение альвеолярно-капиллярной мембраны и легочной ткани может играть важную роль в патогенезе ИПЛ при СЗСТ. При исследовании показателей функции внешнего дыхания больные с СЗСТ демонстрируют снижение диффузионной способности легких (ДСЛ) и рестриктивный тип нарушения вентиляции. Прогноз ИПЛ у пациентов с СЗСТ более благоприятный, чем при РА и ССД. ЛГ является основной причиной смерти больных с СЗСТ [1–3, 14, 15].
Гранулематоз Вегенера (ГВ) — системный некротический васкулит мелких вен и артерий с образованием гранулем в сосудистых стенках и окружающих тканях дыхательных путей, почек и других органов.
ПЛ развивается у большинства пациентов с ГВ. Клинические проявления ПЛ при ГВ разнообразны, начиная от бессимптомных узелков в легких и кончая фульминантным альвеолярным кровотечением. ГВ может сопровождаться образованием опухолевидных инфильтратов с неровными краями, которые могут распадаться и образовывать полости. Плеврит, легочное кровотечение и увеличение лимфатических узлов средостения встречаются редко. Поражение трахеальных или бронхиальных стенок обычно проявляется гранулематозным утолщением слизистой оболочки или подслизистого слоя, при этом возникает обструктивный тип нарушения вентиляции легких. Частое осложнение — коллапс бронхов и постобструктивная пневмония. Инфильтраты, которые могут увеличиваться и уменьшаться, первоначально часто неправильно диагностируются как пневмония. Примерно в 20% случаев развивается прогрессирующая легочная недостаточность, связанная с ЛФ, пневмонией или пневмонитом, индуцированным циклофосфамидом. ДСЛ, как правило, уменьшена, но при развитии диффузных альвеолярных геморрагий наблюдают ее рост. Описаны случаи развития бронхоплевральных свищей [1–3].
Синдром Чарджа–Строса (СЧС) — эозинофильное, гранулематозное воспаление респираторного тракта и некротизирующий васкулит, поражающий мелкие и средние сосуды, часто сочетающийся с астмой и эозинофилией.
Легкие — это наиболее поражаемый орган при данном синдроме; более 90% пациентов с синдромом СЧС в анамнезе имеют астму. При рентгенологическом исследовании легких выявляются очаги консолидации, распределяющиеся по периферии, которые часто бывают преходящими. Могут появляться узелки, при распаде не образующие полости. Другие менее распространенные проявления ПЛ включают утолщение междольковой перегородки и утолщение бронхиальной стенки. Плевральные выпоты образуются редко.
Существуют три фазы развития СЧС: продромальная фаза, которая характеризуется наличием аллергических заболеваний (как правило, астма или аллергический ринит), может продолжаться от нескольких месяцев до многих лет; эозинофилия/фаза инфильтрации тканей, в которой может наблюдаться удивительно высокая периферическая эозинофилия, а также инфильтрация эозинофилами тканей легких, желудочно-кишечного тракта и других органов; васкулитная фаза, в которой некротический васкулит поражает широкий спектр органов — сердце, легкие, периферические нервы и кожу. Диагноз приходится верифицировать с другими васкулитами, в первую очередь ГВ [1–3].
Синдром Гудпасчера (СГ) (геморрагический легочно-почечный синдром) — прогрессирующее аутоиммунное заболевание легких и почек, характеризующееся образованием антител к базальным мембранам капилляров клубочков почек и альвеол и проявляющееся сочетанием легочных и почечных геморрагий.
Патоморфологически в легких наблюдается картина венулитов, артериолитов, капилляритов с выраженными явлениями деструкции и пролиферации; поражение капилляров наблюдается преимущественно в межальвеолярных перегородках, развивается альвеолит с геморрагическим экссудатом в альвеолах.
В большинстве случаев ПЛ и почек происходит одновременно. Клинические проявления ПЛ включают в себя кашель, одышку и кровохарканье, которое может появляться на несколько месяцев раньше признаков поражения почек. В развитии альвеолита при СГ огромное значение имеет активация альвеолярных макрофагов. В активированном состоянии они выделяют около 40 цитокинов. Цитокины I группы (хемотаксины, лейкотриены, интерлейкин-8) усиливают поступление полиморфноядерных лейкоцитов в легкие. Цитокины II группы (факторы роста — тромбоцитарный, макрофагальный) способствуют перемещению в легкие фибробластов. Альвеолярные макрофаги продуцируют также активные формы кислорода, протеазы, повреждающие легочную ткань.
Исследование жидкости бронхоальвеолярного лаважа (ЖБАЛ) не является диагностическим при СГ, но может использоваться для подтверждения наличия диффузной альвеолярной геморрагии у пациентов с гломерулонефритом и легочными инфильтратами, но без кровохарканья. ЖБАЛ, которая остается геморрагической после многократных промываний, позволяет подтвердить диффузный геморрагический синдром, особенно при сопутствующем снижении гематокрита.
Гистологическое и иммунологическое исследование биоптатов легочной ткани при СГ характеризуется признаками геморрагического альвеолита, гемосидероза и интерстициального фиброза, а также линейных отложений иммуноглобулина G (IgG) и С3-компонента комплемента на базальных мембранах легочных альвеол.
Рентгено-компьютерное исследование легких при СГ демонстрирует наличие легочных инфильтратов в прикорневой области с распространением на нижние и средние отделы легких. Исследование легочных тестов выявляет рестриктивный тип нарушения вентиляции легких (снижение жизненной емкости легких — ЖЕЛ), но по мере прогрессирования заболевания присоединяются обструктивные изменения (снижение объема форсированного выдоха за 1 сек — ОФВ1, индекса Тиффно) [1–3].
Болезнь Бехтерева (ББ, анкилозирующий спондилит, АС) — хроническое системное заболевание, характеризующееся воспалительным поражением суставов позвоночника, околопозвоночных тканей и крестцово-подвздошных сочленений с анкилозированием межпозвоночных суставов и развитием кальцификации спинальных связок.
ПЛ у больных АС встречается в 50–85% случаев и обусловлено анкилозирующим процессом в грудном отделе позвоночника, снижением дыхательной экскурсий грудной клетки, утомлением и слабостью дыхательных мышц. У больных АС чаще всего развивается эмфизема легких, затем ИПЛ, хроническая обструктивная болезнь легких (ХОБЛ), апикальный фиброз, бронхоэктазия и поражение плевры. Апикальный пневмофиброз, который встречается нечасто (3–4%), требует проведения дифференциальной диагностики с туберкулезными изменениями. Фиброз верхней доли легкого обычно протекает бессимптомно, но может вызывать кашель, отделение мокроты и одышку.
При АС чаще встречается рестриктивный тип нарушения вентиляции легких. У больных АС с хронической обструктивной болезнью легких (ХОБЛ) исследование ФВД демонстрирует обструктивные вентиляционные изменения [1, 16, 17].
Таким образом, при САЗ могут наблюдаться различные типы легочной патологии. Развитие ПЛ обусловлено особенностями патофизиологических характеристик основного заболевания. Основные легочные проявления САЗ включают заболевания плевры, ИПЛ, поражение бронхиального дерева. При РА и СКВ чаще, чем при других САЗ, встречается поражение плевры. ИПЛ в настоящее время все больше признается как самое частое и серьезное проявление САЗ. ПЛ у больных с САЗ оказывает существенное негативное воздействие на качество жизни (КЖ): у больных снижаются показатели КЖ, характеризующие физический, психоэмоциональный статус и социальную активность.
ПЛ при САЗ имеет большое значение в формировании облика заболевания, при этом во многом определяет его тяжесть и прогноз. Наряду с базисной терапией САЗ, ПЛ необходимо рассматривать как важную мишень для терапевтического воздействия.
Литература
- Ревматология, национальное руководство. Под ред. Е. Л. Насонова, В. А. Насоновой. М.: ГЭОТАР-Медиа, 2008.
- Castelino F. V., Varga J. Interstitial lung disease in connective tissue diseases: evolving concepts of pathogenesis and management // Arthritis Research & Therapy. 2010; 12: 213.
- Bouros D., Pneumatikos I., Tzouvelekis A. Pleural involvement in systemic autoimmune disorders // Respiration. 2008; 75: 361–371.
- Kriegel M. A., Van Beek C., Mostaghimi A. et al. Sterile empyematous pleural effusion in a patient with systemic lupus erythematosus: a diagnostic challenge // Lupus. 2009; 18: 581–585.
- Pego-Reigosa J. M., Medeiros D. A., Osenberg D. A. Respiratory manifestations of systemic lupus erythematosus: old and new concepts // Best Pract Res Clin Rheumatol. 2009; 23: 460–480.
- Varda J., Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder // J Clin Invest. 2007; 117: 557–567.
- Yanaba K., Hasegawa M., Takehara K. et al. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis // J Rheumatol. 2004; 31: 1112–1120.
- McNearney T. A., Revelle J. D., Fischbach M. et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors // Arthritis Rheum. 2007; 57: 318–326.
- Tillie-Leblond I., Wislez M., Valeyre D. et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset // Thorax. 2008; 63: 53–59.
- Chen I. J., Jan Wu Y. J., Lin C. W. et al. Interstitial lung disease in polymyositis and dermatomyositis // Clin Rheumatol. 2009; 28: 639–646.
- Fujisawa T., Suda T., Nakamura Y. et al. Differences in clinical features and prognosis of interstitial lung diseases between polymyositis and dermatomyositis // J Rheumatol. 2005; 32: 58–64.
- Ito I., Nagai S., Kitaichi M. et al. Pulmonary manifestations of primary Sjögren’s syndrome: a clinical, radiologic, and pathologic study // Am J Respir Crit Care Med. 2005; 171: 632–638.
- Parambil J. G., Myers J. L., Lindell R. M. et al. Interstitial lung disease in primary Sjögren’s syndrome // Chest. 2006; 130: 1489–1495.
- Bodolay E., Szekanecz Z., Devenyi K. et al. Evaluation of interstitial lung disease in mixed connective tissue disease (MCTD) // Rheumatol. 2005; 44: 656–661.
- Kinder B. W., Shariat C., Collard H. R. et al. Undifferentiated connective tissue disease-associated interstitial lung disease: changes in lung function // Lung 2010; 188: 143–149
- Quismorio F. P. Jr. Pulmonary involvement in ankylosing spondylitis // Curr Opin Pulm Med. 2006; 12: 342–345.
- Lee C. C., Lee S. H., Chang I. J. et al. Spontaneous pneumothorax associated with ankylosis spondylitis // Rheumatol. 2005; 44: 1538–1541.
Д. В. Бестаев 1 , кандидат медицинских наук
Е. Л. Насонов, доктор медицинских наук, профессор, академик РАН
ФГБУ НИИР им. В. А. Насоновой РАМН, Москва
Ревматоидный артрит (РА) – часто встречающаяся патология (8-10 случаев из 1000). Однако существует ряд других системных соединительнотканных болезней, которые имеют сходные симптомы с РА. Процесс выявления заболевания из группы подобных называется дифференциальной диагностикой.
Болезни со сходной симптоматикой
Группа аутоиммунных заболеваний соединительной ткани, с которыми проводится дифференциальный диагноз при ревматоидном артрите, включает:
- деформирующий остеоартроз с реактивным синовиитом;
- ревматизм;
- подагра;
- системная красная волчанка;
- псориатический артрит;
- инфекционные (вирусные) артриты;
- системная склеродермия;
- анкилозирующий спондилоартрит;
- реактивные артриты.
Эти патологические процессы, как и тот, с которыми необходимо их дифференцировать, в одну из своих фаз характеризуются острым воспалением суставных и околосуставных тканей, поэтому между ними проводится дифференциальная диагностика артритов.
Диагностика

Для подтверждения или опровержения диагноза следует учитывать следующие критерии:
- поражение костных сочленений симметрично: если страдают суставы левой руки, страдают аналогичные правой руки;
- изменения необратимы: измененная в результате разрастания пануса форма суставов остается такой пожизненно;
- утренняя скованность в суставах составляет более получаса;
- характерно поражение:
- ІІ и ІІІ пальцев обеих рук в области проксимальных межфаланговых и пястно-фаланговых сочленений;
- запястий и колен;
- голеностопных суставов и локтей.
- наличие внесуставных проявлений:
- ревматоидные узелки;
- лимфаденопатия;
- висцериты.
- в анализе крови:
- наличие изменений, характерных для любых воспалительных процессов в организме;
- положительная реакция на ревматоидный фактор (HLA-B27).
- на рентген-снимке:
- эпифизарный остеопороз;
- эрозивный артрит;
- сужение суставных щелей.
На основе этих признаков проводится дифференциальная диагностика ревматоидного артрита с другими болезнями из его группы.
Описание артритов для дифференцированного диагноза с ревматоидным артритом

Для ревматизма характерно не прогрессирующее развитие артрита, а очень частые рецидивы с межприступными паузами различной длительности. Сочленения верхних конечностей поражаются несимметрично; возможен как моно- (один), так и олиго- (несколько) артрит. Околосуставные ткани припухлые, кожа над ними синюшного оттенка. Суставные изменения обратимы и исчезают в течение 2-3 дней, вместе с окончанием периода обострения.
Поражены мелкие суставы; все деформации обратимы. На фоне других симптомов:
Артритические проявления отходят на второй план.
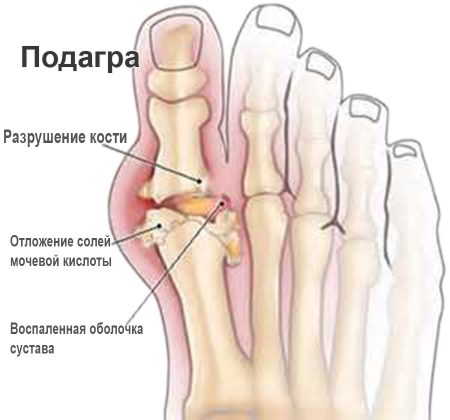
Характерная локализация для подагры – плюснефаланговый сустав І пальца. Кожа над ним раскрасневшаяся, при движении боль усиливается. Если не начать лечение своевременно, вскоре вовлекаются в патологический процесс другие суставы — симметрично или асимметрично. В суставных сумках могут прощупываться тофусы (узелки) различной величины.
В крови повышена концентрация мочевой кислоты. При отсутствии адекватного лечения и изменения образа жизни вскоре нарушается функция почек. Изменения обратимы: первоначальное функционирование и форма суставов возвращаются с выведением уратов.
За счет отека суставы приобретают форму веретена, а кожа над ними становится малинового или синюшно-багрового цвета. Помимо суставных проявлений при ПА на коже появляются псориатические бляшки, страдают производные кожи: слоятся ногти, появляется перхоть, могут выпадать волосы. Положителен анализ крови на ревматоидный фактор, присутствует рентгенологически-выраженная деструкция костной ткани.
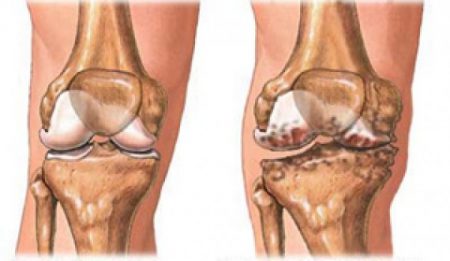
Нарушается форма дистальных межфаланговых суставов кистей, а также мелких и крупных костных соединений нижних конечностей. Боль механического типа: не проявляется в покое и усиливается с увеличением интенсивности движений. Но при этом отсутствуют значительные нарушения их функций, за исключением тазобедренного сочленения.
Висцеропатии не характерны. Данные анализа крови соответствуют наличию в организме слабой воспалительной реакции, а то и вовсе норме. На рентгенологическом снимке – субхондральный (подхрящевой) остеосклероз, а также значительные костные выросты – проявления остеофитоза.
Имеется временная связь с перенесенным инфекционным заболеванием мочеполовой системы или кишечника – либо недолеченным, либо вообще невыявленным (вариант не составит труда установить по совокупности анализов крови, мочи, соскоба из уретры и копрограммы). Артрит возникает в течение полугода после занесения инфекции. Ревматический фактор положительный. Изменения в суставах обратимы.
Проявляется мышечно-суставным синдромом: возникает симметричный полиартрит и периартрит, в результате наблюдаются сгибательные контрактуры. Все это сопровождается миозитом и псориатическим поражением кожи – плотным отеком лица и кистей, затрудняющим движения, в том числе и мимических мышц (возникает симптом маскообразного лица). Такие же фиброзные изменения происходят во внутренних органах: ЖК тракта, почках, кровеносных сосудах и др. В крови повышено содержание гамма-глобулина.
Поражение суставов асимметрично, в основном страдают мелкие сочленения. Воспаляются суставные связки, околосуставные ткани. Лечение основного заболевания – антибактериальная или противовирусная терапия – быстро устраняет суставные проявления.
Методы исследования, которые необходимо провести для подтверждения или опровержения диагноза:
- общий анализ крови (на выявление воспалительных явлений: повышения количества лейкоцитов и сдвига лейкоцитарной формулы);
- биохимический анализ крови (на наличие ревматического фактора HLA-B27);
- анализ мочи и копрограмма (на выявление инфекции или ее последствий);
- рентгенологический снимок беспокоящих костных соединений (для выявления наличия/отсутствия выростов, фиброзных процессов, остеопороза и др.).
О пределение: Гетерогенные нарушения, имеющие определенные общие черты, включая воспалительные процессы в коже, суставах и других структурах, богатых соединительной тканью, а также общие черты нарушения иммунной регуляции с образованием ауто-антител и нарушением клеточно-опосредованного иммунитета. Несмотря на то, что установлены определенные различия сущности болезней, их проявления могут значительно варьировать у разных больных, они могут частично перекрывать специфические клинические признаки отдельных болезней.
Системная красная волчанка
Определение, патогенез: Системная красная волчанка (СКВ) — заболевание неизвестной этиологии, при котором ткани и клетки повреждаются вследствие отложения в них патогенных антител и иммунных комплексов. Условия окружающей среды, генетические и половые гормональные факторы играют роль в патогенезе болезни. Отмечается гиперактивность В-лимфоцитов, образование специфических аутоантител к нуклеарным антигенным детерминантам и нарушение функции Т-лимфоцитов.
Диагностика: Включает анамнез и физическое обследование, рентгенологические исследования, ЭКГ, общий анализ мочи, общий анализ крови, СОЭ, определение в сыворотке крови уровней Ig, AHA и их подтипов (двухспиральная ДНК, односпиральная ДНК, антитела к гладким мышцам, антигистоны), а также уровней комплемента (СЗ, С4, СН50), пробы на сифилис, ПВ и ЧТВ.
Диагноз достоверен при наличии четырех или более критериев (см. Table 284-3,р. 1645 in HPIM-13).
Направлено на ограничение воспалительного процесса. Применяют салицилаты (особенно аспирин, покрытый оболочкой, растворимой в кишечнике, 2-4 г в день в нескольких дозах), НПВС (ибупрофен 400-800 мг 3-4 раза в день) и гидрокси-хлорохин, 400 мг в день; глюкокортикоиды (преднизон 1-2 мг/(кг х день) или его эквиваленты необходимы при состояниях, угрожающих жизни, или при тяжелых проявлениях заболевания; цитостатические препараты циклофосфамид 1,5-2,5 мг/ (кг х день) или азатиоприн 2-3 мг/(кг х день) применяют в случаях, когда стероиды не обеспечивают подавление симптомов заболевания; внутривенное введение пульс-доз циклофосфамида (10-15 мг/кг каждые 4 нед) — альтернатива ежедневному приему цитоксических средств.
Ревматоидный артрит
Определение и патогенез: Ревматоидный артрит (РА) — хроническое системное заболевание неизвестной этиологии, характеризующееся главным образом персистирующим воспалительным синовитом, симметрично поражающим периферические суставы. Хрящевая деструкция, костные эрозии и деформация суставов служат критериями персистирую-щего синовиального воспаления. Патогенез заболевания недостаточно ясен; наблюдают синовиальную гиперплазию, лимфоцитарную инфильтрацию синовиальной ткани, инфильтрацию суставов нейтрофилами, выделение протеаз и активацию хон-дроцитов.
Диагностика: Анамнез и физическое обследование с тщательным исследованием всех суставов; общий анализ крови, СОЭ, РФ, уровни комплемента, анализ синовиальной жидкости, рентгенологическое исследование грудной клетки и суставов.
Диагностика не представляет трудностей у больных с типичной клинической картиной заболевания. В ранней стадии болезни диагноз может быть не ясен. Новые критерии более чувствительны и специфичны (см. Table 285-3, р. 1653, HPIM-13).
1) основными средствами служат: аспирин (2-4 г в день в несколько приемов) и НПВС (ибупрофен 400-800 мг 3-4 раза в день); 2) прием солей золота (аурано-фин 6 мг в день внутрь), гидроксихлорохин 400 мг в день, D-пеницилламин 500 мг в день, сульфасалазин 500 мг в день; 3) преднизон, в малой дозировке (7,5 мгв день); 4) метотрексат 7,5-15 мг в неделю; 5) оперативное лечение.
Системная склеродермия
Определение и патогенез: Системное нарушение, характеризующееся воспалительными, сосудистыми и фиброзными изменениями кожи и различных систем внутренних органов (главным образом ЖКТ, легких, сердца и почек). Патогенез не известен; в результате патологического процесса происходит повреждение эндотелиальных клеток и, в конечном итоге, пролиферация интимы, фиброз и сосудистая облитерация.
Клинические проявления: Феномен Рейно, фиброз кожи (склеродермия), телеангиэктазии, кальциноз, снижение перистальтики пищевода, артралгии и (или) артрит, гипофункция кишечника, пневмофиброз, артериальная гипертензия, почечная недостаточность (ведущая причина смерти).
Диагностика: Анамнез и физическое обследование, СОЭ, рентгенография грудной клетки, контрастирование пищевода барием, AHA (специфические антитела включают антитела к ядерным антигенам, рибонуклеопротеину, центромеру и антитопоизомера-зе), ЭКГ, общий анализ мочи, биопсия кожи.
Радикального лечения склеродермии нет, но адекватная терапия может облегчить симптомы и улучшить состояние больного. D-пеницилламин в начальной дозе 250 мг в день может уменьшить уплотнение кожи и предотвратить поражение внутренних органов. Глюкокортикоиды (преднизон 40-60 мг в день, с последующим снижением дозы) показаны при миозите или перикардите. Блокаторы кальциевых каналов (нифедипин 10-20 мг внутрь 3 раза в день) показаны при феномене Рейно, кроме того, помогает ношение теплой одежды, прекращение курения. Антациды, Н2-антагонисты (ранитидин 150 мг внутрь 2 раза в день), омепразол 20-40 мг внутрь перед сном и метоклопрамид 10 мг внутрь, 2-3 раза в день — при пищеводном реф-люксе. Ингибиторы АПФ (каптоприл 25 мг внутрь 3 раза в день) эффективны в лечении артериальной гипертензии и прогрессирования болезни почек.
Смешанное заболевание соединительной ткани
Определение и патогенез: Синдром, проявляющийся сочетанием клинических проявлений, характерных для СКВ, склеродермии, полимиозита и РА; выявляются необычно высокие титры циркулирующих антител к ядерному рибонуклеопротеину.
Механизмы патогенеза не известны, но очевидны нарушения иммунорегуляции и пролиферативные поражения интимы и (или) средней оболочки сосудов, ведущие к сужению просвета сосудов.
Клинические проявления: Феномен Рейно, полиартрит, припухлость кистей рук или склеродактилия, дисфункция пищевода, пневмофиброз, миозит. Вовлечение в патологический процесс почек является менее типичным, чем при склеродермии. Лабораторные данные включают высокие титры AHA, положительный РФ у 50 % больных, очень высокие титры антител к рибонуклеопротеиновому компоненту экстрагированного ядернот го антигена.
Диагностика: Как при склеродермии и СКВ.
Подобно лечению при СКВ: в основном, направлено на подавление воспалительных проявлений заболевания.
Синдром Шегрена
Определение и патогенез: Иммунологическое нарушение, характеризующееся прогрессирующей деструкцией экзокринных желез, ведущей к сухости слизистых и конъюнктивальных оболочек (сухой синдром); может быть первичным или сочетается с другими аутоиммунными болезнями; в пораженных тканях отмечают лимфоцитарную инфильтрацию и отложение иммунных комплексов.
Клинические проявления: Ксеростомия и сухой кератоконъюнктивит, нефрит, васкулит (обычно кожный), полиневропатия, интерстициальный пневмонит, псевдолимфома, аутоиммунный тиреоидит, врожденные дефекты проводящей системы сердца у детей, рожденных женщинами с антителами к односпиральной ДНК.
Диагностика: Анамнез и физическое обследование, специальное внимание уделяют определению распространенности поражений и наличию других аутоиммунных нарушений; общий анализ крови, общий анализ мочи, рентгенография грудной клетки, ЭКГ, функции щитовидной железы, тест Ширмера, СОЭ, криоглобулин.
Симптоматическое облегчение сухости с помощью искусственных слез, глазных увлажняющих мазей, закапывания в носовые ходы изотонического раствора, частые питье воды, увлажняющие примочки на кожу; лечение аутоиммунных проявлений. Бромгексин, 48 мг в день, уменьшает проявления сухости. Гидроксихлорохин, 200 мг в день, частично корректирует гипергаммаглобулинемию, образование аутоантител и СОЭ. Глюкокортикоиды или другие иммунодепрессанты показаны при экстрагландулярных проявлениях.
Читайте также: