
Заболевания связанные с нарушением обмена соединительной ткани
Наследственные болезни СТ относятся к наиболее распространенным генетическим синдромам. СТ является одной из наиболее распространенных тканевых структур в организме человека. Функции ее многообразны.
Наиболее важными из них являются:
1) Опорная, определяемая в первую очередь коллагеновыми волокнами.
1)Защитная или барьерная, включающая не только механическую, но и элементы имунной защиты, синтез веществ с антимикробным и антивирусным действием.
2)Метаболическая или трофическая, так как именно СТ осуществляет активный обмен между кровью и органами, участвует в регуляции обменных процессов, синтезируя и секретируя цитокины, ферменты, простагландины, факторы роста и др.
3)Морфологическая или структурообразовательная функция, так как СТ оказывает регулирующее действие на размножение и дифференциацию некоторых типов клеток и формирование структур органов в эмбриональном и постнатальном периоде.
4)Пластическая или репаративная функция, связанная с выраженными регенераторными свойствами фибробластов.
Основными клетками СТ являются фибробласты, синтезирующие и формирующие основное вещество, фибриллярные белки - коллаген и эластин. Кроме того, в СТ имеются тучные, ретикулярные, жировые, пигментные и эндотелиальные клетки, а также макрофаги, лейкоциты и плазматические клетки. Внеклеточные элементы СТ состоят из фибриллярных структур (коллагена и эластина) и основного вещества. Основное вещество состоит главным образом из высокомолекулярных протеогликанов и гликопротеидов.
Существенным химическим компонентом СТ являются мукополисахариды. Помимо структурной функции, они обеспечивают сохранение постоянной проницаемости ткани и водно-солевое равновесие, выполняют защитные функции в составе секретов слизистых желез.
Наибольшее физиологическое значение имеют так называемые кислые мукополисахариды (кислые гликозоаминогликаны), подразделяемые на две группы: сульфированные (хондроитинсульфат А, В, С, гепарансульфат, кератосульфат) и несульфированные: гиалуроновая кислота , хондроитин. Хондроитинсульфаты в наибольшем количестве содержатся в хрящах (40%) веса.
Гепаритинсульфат- в печени, легких, стенке аорты.
Хондроитин- в роговице глаза.
Гиалуроновая кислота связывает клетки друг с другом, играет роль барьера для микроорганизмов. Особенно много гиалуроновой кислоты в почках, синовиальной жидкости, стекловидном теле.
Дерматансульфат в основном содержится в коже, связках, сухожилиях, легких и склере.
Кератосульфат- в роговице, хряще, кости.
Волокна СТ бывают двух типов: коллагеновые и эластические, причем коллагеновых волокон имеется 4 разновидности. В тканях до 90% волокон представлены коллагеном 1 типа.
Классификация нарушений обмена соединительной ткани.
Систематизация болезней СТ основана на клинических проявлениях. Однако следует отметить, что классификация осложняется большой клинической гетерогенностью этих заболеваний. Поэтому принято выделять синдромы с четко очерченной клиникой болезни (дифференцированные формы) и состояния, имеющие признаки поражения СТ без возможности четкой дифференцировки (недифференцированные соединительнотканные дисплазии).
Возникают при нарушении синтеза и утилизации гликозоаминогликанов, которые накапливаются в лизосомах клеток всех тканей организма больного.
Различают 7 типов заболевания в зависимости от дефекта фермента в цепи обмена гликозоаминогликанов.
Общие клинические признаки:
1.Поражение скелета: низкий рост, костные деформации, контрактуры суставов
2.Формирование необычного лица
4.Часто развивается умственная отсталость, глухота.
Название болезни произошло от французского слова Gargoillе, так как внешний вид больных напоминал своеобразные уродливые фигуры, украшающие собор Нотр-Дам в Париже.
Первое описание болезни было дано Hunter в 1917 году.
Мукополисахаридоз, тип I (синдром Гурлера)
Впервые описан в 1919 г. G.Hurler.
Популяционная частота - не известна. Соотношение полов 1:1.
Тип наследования: аутоссомно-рецессивный.
1.Задержка роста. Дети рождаются без внешних изменений, иногда с большой массой тела. Рост замедляется после первого года жизни.
2.Выраженная умственная отсталость. Психомоторное развитие протекает нормально до 2 лет. Для поздних стадий заболевания характерна глубокая деменция.
3.Характерные черепно-лицевые дисморфии, множественный дизостоз.В первые месяцы жизни черты лица становятся грубыми, характерны запавшая переносица, тугоподвижность суставов, тораколюмбальный кифоз. Рот обычно открыт. Постоянными симптомами, развивающимися на втором году жизни являются короткая шея, воронкообразная или килевидная грудная клетка, паховые и пупочные грыжи. Гипертрихоз,. Особенно на разгибательных поверхностях конечностей и на спине, макроцефалия, увеличение языка и губ, мелкие редко посаженные зубы,ринит, шумное дыхание, ограничение подвижности в межфаланговых, локтевых, плечевых, тазобедренных суставах. Кожа сухая, грубая, бледная. Гепатоспленомегалия. Кардиомегалия. Глухота.
4.Помутнение роговицы, слепота.
1.Рентгенологически: уплощение и расширение турецкого седла, расширение диафизов трубчатых костей, особенно верхних конечностей и ребер, клювовидная форма тел позвонков.
2.УЗИ внутренних органов
3.Определение экскреции гликозоаминогликанов в суточной моче и разделение их на фракции (повышена экскреция дерматансульфата и гепарансульфата)
4.Энзимодиагностика пораженного фермента в печени (дефицит L- идуронидазы)
5.Больные погибают в возрасте до 10 лет от бронхолегочных инфекций и сердечной декомпенсации. На аутопсии обнаруживают отложение мукополисахаридов в клапанах сердца, в хрящевых клетках, в периферических нервных ганглиях, сетчатке, склере, роговице, почках, селезенке.
мукополисахаридозы - группа заболеваний соединительной ткани, связанных с нарушением обмена кислых гликозаминогликанов.
Фенилкетонурия - заболевание, обусловленное недостаточной (отсутствием или снижением) активностью фенилаланингидроксилазы. Средняя частота ФКУ составляет 1:10000. Ген, кодирующий ФКУ, локализован в длинном плече 12-й хромосомы.
В основе патогенеза ФКУ - накопление фенилаланина и продуктов побочных реакций - фенилпировиноградной, фенилуксусной и фенилмолочной кислот оказывающих токсическое действие на мозг и клетки внутренних органов больного.
Клинические проявления по мере накопления фенилаланина нарастают, и к шестимесячному возрасту у ребенка формируются необратимые изменения ЦНС и других органов и систем (дети с ФКУ склонны к дерматитам). Для больных ФКУ характерна прогрессирующая умственная отсталость, эписиндром (причем у 40% больных наблюдаются эпиприступы.
Причиной атипичной ФКУ или ФКУ с легким течением может быть гетерозиготность по мутациям, полученным больным ребенком от родителей.
Муковисцидоз — генетическое заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза.
Уже в периоде новорожденности и грудном возрасте возникает кашель, приступы удушья, одышка, иногда рвота. Периодически возникает мучительный сильный кашель, особенно по ночам. Мокрота вязкая, иногда гнойная.
Так как поражаются все органы, содержащие слизеобразующие железы, типичны колитический синдром, хронический холецистит, синуситы.
Синдром гипотиреоза – клинический синдром, вызванный длительным, стойким недостатком гормонов щитовидной железы в организме или снижением их биологического эффекта на тканевом уровне. Врожденный гипотиреоз наблюдается с частотой 1 случай на 4000-5000 родов.
Клиника: Взрослые обычно жалуются на слабость, повышенную утомляемость, плохую переносимость холода, увеличение массы тела при низком аппетите, выпадение волос, снижение памяти, миалгии, артралгии. При осмотре кожа отечная, бледная, сухая, холодная на ощупь. Она может быть желтоватой с оранжевым оттенком в результате отложения каротина. Лицо одутловатое, мимика скудная, глазные щели сужены за счет отека век. Заметно шелушение кожи, выпадение волос. В результате отека голосовых связок голос становится более хриплым. Нередко наблюдается склонность к инфекциям верхних дыхательных путей.
При гипотиреозе часто поражается сердечно-сосудистая система и развивается брадикардия, артериальная гипотензия. Часто встречаются изменения желудочно-кишечного тракта, склонность к запорам, метеоризм в связи с ослаблением двигательной функции кишечника.
По мере прогрессирования заболевания нарастают расстройства центральной нервной системы (ЦНС) - вялость, апатия, сонливость, нарушение психики вплоть до психоза с депрессией и т.д.
Характерно нарушение функции половой системы - снижение либидо у мужчин, нерегулярный менструальный цикл у женщин.
Выраженный гипотиреоз (микседема) с массивным отложением мукополисахаридов в ткани железы обычно бывает идиопатическим и развивается постепенно. При микседеме особенно заметны перечисленные симптомы, в частности изменения кожи, брадикардия, накопление жидкости в полостях, увеличение языка в размерах, изменения ЦНС.
Ахондроплазия - приблизительно 1 случай на 26-40тыс. новорожденных. Является наиболее распространенной формой карликовости, около 70% всех случаев карликовости.
Клинически врождённое укорочение конечностей с низким ростом (средний рост мужчин -131 см; женщин - 124 см), ожирение, аномальная форма черепа (нависающий лоб, относительное увеличение мозгового черепа, гипоплазия средней трети лица с глубокой переносицей), косоглазие, кондуктивная тугоухость, дыхательная недостаточность , поясничный горб, выраженный поясничный лордоз, ограничение разгибания в локтевых и коленных суставах, широкая кисть типа трезубца, брахидактилия, искривление ног, гидроцефалия, часто сужение спинномозгового канала, особенно в поясничном отделе.
Синдром Марфана системное поражение соединительной ткани, проявляющееся «патологическими изменениями опорно-двигательного аппарата, глаз и сердечно-сосудистой системы. Впервые синдром описал Марфан в 1896 г. Встречается редко.
Заболевание наследуется по А-Д типу. Установлено, что при С.М. основной дефект связан с нарушениями коллагена, хотя не исключена возможность поражения эластических волокон соединительной ткани.
Клиника: у больных резко выражен астенический тип сложения (высокий рост, истончение подкожной клетчатки, мышечная слабость), удлинение пальцев кистей и стоп, долихоцефалия — изменение формы головы, когда продольный размер значительно превышает поперечный, так называемое птичье лицо — узкое, с близкорасположенными глазами, тонким носом и выступающей вперед верхней челюстью (прогнатия); деформация ушных раковин, высокое небо. Иногда наблюдается расщепление твердого неба (волчья пасть). Конечности, пальцы кистей и стоп удлинены, грудная клетка воронкообразной или килевидной формы, ребра тонкие и длинные, межреберные промежутки широкие, позвоночник искривлен. Отмечаются разболтанность суставов, иногда с переразгибанием в коленных суставах, плоскостопие.
Из поражений глаз наиболее часто встречается вывих или подвывих хрусталика, обусловливающий высокую степень миопии. Нередко обнаруживают отсутствие ресниц, голубую окраску склер, косоглазие, катаракту , дистрофические изменения сетчатки глаза.
У больных с М.с. отмечаются признаки вегетативно-сосудистой дистонии: потливость, похолодание конечностей, мраморный рисунок кожи, акроцианоз. При исследовании сердца часто диагностируют изменения в проводящей системе, метаболические нарушения в миокарде, расширение просвета аорты и др. Нередко формируется аневризма аорты или легочного ствола.
Синдром Вильсона-Коновалова составляет от 0,6 до 3 больных на 100000 населения.
В основе клинических проявлений лежит нарушение обмена меди в организме. В норме основная масса меди после всасывания в кишечнике выводится с желчью и мочой. При нарушение транспорта меди увеличивается вывод меди через почки и, соответственно, уменьшается ее содержание в крови. Это приводит к нарушению обмена кислорода крови, поскольку медь входит в состав специфических ферментов крови, ответственных за газообмен организма. В итоге, из-за нарушения обмена меди, она накапливается в печени, почках, роговице глаза и головном мозге. В печени из-за этого развивается цирроз, накопление шлаков в организме.
Накопление меди в почках сопровождается развитием неврита и гломерулонефрита - т.е. поражением почечных клубочков и канальцев; это приводит к нарушению фильтрации ионов и белка, их повышенному выведению с мочой из организма.
Накопление меди в роговице глаза не вызывает нарушения зрения , однако приводит к появлению специфического диагностического симптома этой болезни - желтоватого цвета кругов на радужке, которые видны невооруженным глазом ( сочетание наличия медных роговичных колец Кайзера-Флейшера с резким снижением уровня в сыворотке крови основного медьсодержащего белка церулоплазмина (ЦП).
Накопление меди в головном мозге, приводит к развитию ряда нервно-психических симптомов.
При Х-сцепленном типе наследования мутантный ген расположен в X-хромосоме. Если при этом мутация обладает доминантным эффектом, то больными могут быть как мужчины, так и женщины.
К доминантному, сцепленному с Х-хромосомой, типу наследования относится известная детским врачам патология витамин Д-резистентный рахит. Диагноз этого тяжелейшего рахита, который не проходит под воздействием больших доз витамина Д, подтверждается наличием подобного заболевания у части родственников как мужского, так и женского пола.
Наиболее известными Х-сцепленными заболеваниями являются гемофилии А и В, а так же тяжелейшая патология мышечной системы - миодистрофия Дюшенна Беккера. В основе развития гемофилии А лежат мутации гена, ответственного за синтез VIII фактора свертываемости крови, а при гемофилии В дефектным оказывается IX фактор свертываемости крови.
Биохимические методы
Эти методы помогают обнаружить целый ряд заболеваний с нарушениями обмена веществ . Исследованию подлежат кровь, моча, пунктаты костного мозга, амниотическая жидкость, сперма, пот, волосы, ногти, кал и др.
ХРОМАТОГРАФИЯ (от греч. chroma - род. п. chromatos - цвет и . графия), метод разделения и анализа смесей, основан на различном распределении их компонентов между двумя фазами - неподвижной и подвижной (элюентом). Хроматография может быть основана на различной способности компонентов к адсорбции (адсорбционная хроматография), абсорбции (распределительная хроматография), ионному обмену (ионообменная хроматография) или др.
Микробиологический ингибиторный тест Гатри позволяет выявлять некоторые биохимические нарушения у новорожденных. Из пятки новорожденного берут каплю крови на диски фильтровальной бумаги, которые помещают на агаровую культуру. Последнюю выращивают на минимальной питательной среде, содержащей антиметаболит искомой аминокислоты (например, фенилаланина). Антиметаболит должен одновременно тормозить рост микроба. При наличии в крови младенца большого количества фенилаланина антиметаболит разрушается и микробы начинают бурно расти. Меняя антиметаболиты, можно диагностировать наличие в крови определенных аминокислот и углеводов (лейцина,гистидина, фруктозы, галактозы и др.).


К настоящему времени описано множество глазных изменений при различных видах нарушений обмена веществ. Необходима- отметить, что иногда именно глазные симптомы являются наиболее ранними признаками патологии обменных процессов в организме.
Своевременное выявление этих симптомов позволяет рано начать лечение и правильно выбрать метод для его осуществления. В связи с этим знание и умение обнаружить первые офтальмологические признаки является насущной необходимостью. Эта тем более важно, что диагностика болезней обмена у детей трудна ввиду их исключительного полиморфизма.
Изменения органа зрения при нарушениях обмена веществ обусловлены сложным процессом обмена в тканях глаза. Несовершество и незрелость ферментативных процессов у детей, отсутствие и недостаточность некоторых элементов, а также их накопление вызывают необратимые изменения в прозрачных средах глаза, сетчатке и других отделах глазного яблока.
Так, структура хрусталика и сетчатки, их метаболизм обусловливаются одновременным воздействием по крайней мере нескольких тысяч химических реакций, катализируемых различными ферментами. Основным источником энергии является глюкоза.
Нарушение обмена глюкозы приводит к накоплению органических кислот и других продуктов метаболизма, которые вызывают грубые изменения в тканях. Вследствие этого, например, при непереносимости галактозы отмечается катарактогенный эффект и появляются дегенеративные изменения в сетчатке.
Белки хрусталика органоспецифичны. Они синтезируются из аминокислот, которые проникают в него через капсулу хрусталика из влаги камеры.
Химическая энергия в виде АТФ в сетчатке образуется в основном за счет гликолиза и дыхания. Сетчатка, так же как и мозг, содержит большое количество глютаминовой кислоты, глютамина и микроэлементов, которые являются компонентом окислительных ферментов.
БОЛЕЗНИ СОЕДИНИТЕЛЬНОЙ ТКАНИ
Наиболее часто морфологической основой развития ряда наследственных болезней является поражение соединительной тканей. Наследственные болезни соединительной ткани мало изучены, дифференциальная диагностика их сложна, особенно у детей раннего возраста при приобретенных клинически сходных (фенотипические) заболеваниях. Для наследственных нарушений метаболизма соединительной ткани характерно поражение органа зрения.
Глазные симптомы могут быть ранними и единственными признаками наследственной патологии соединительной ткани, поэтому наряду с генеалогическими и биохимическими исследованиями при этих заболеваниях важное значение имеют офтальмологические методы диагностики.
Принято различать две основные группы болезней соединительной ткани: к первой группе относятся генетически обусловленные заболевания — мукополисахаридозы, синдром Марфана, гомоцистинурия и др., ко второй — болезни, в развитии которых определенную роль играют такие наследственные факторы, ревматизм, инфекционный неспецифический полиартрит, красная волчанка.
При патологических состояниях соединительной ткани первыми и самыми ранними признаками являются изменения в обмене кислых гликозаминогликанов, что свидетельствует о большой чувствительности этой ткани, в частности межуточного основного вещества, к обменным нарушениям в организме.
Особенно значительные изменения глаз отмечаются при наиболее тяжелой форме наследственного поражения соединительной ткани мукополисахаридозе. Заболевание называют также множественным дизостозом, липохондродистрофией, номедистрофией, остеохондродистрофией.
Наиболее тяжелым заболеванием является мукополисахаридоз I типа — синдром Гурлер, прототип всех мукополисахаридозов, характеризующийся прогрессирующим течением, а также тем, что больные умирают в ранние сроки (до 10—12 лет). Остальные шесть типов мукополисахаридоз а существенно отличаются от первого.
Мукополисахаридоз II типа - синдром Гунтера протекает менее тяжело. Ведущими симптомами являются изменения в костно- суставной системе, но менее выраженные, чем при I типе, а также снижение слуха.
Значительно отличается от первых двух типов мукополисахаридозов III тип - синдром Санфилиппо. Заболевание характеризуется наличием умственной отсталости на фоне незначительных изменений скелета и соматических нарушений.
В клинической картине мукополисахаридоза IV типа — синдрома Моркио на первое место выступают поражения костно-суставной системы деформации грудной клетки и черепа.
Мукополисахаридоз V типа — синдром Шейе — характеризуется почти полной сохранностью интеллекта, преимущественным поражением мелких суставов и умеренно выраженными соматическими изменениями.
Для мукополисахаридоза VI типа — синдрома Марото - Лами характерны грубые деформации скелета при сохранении нормального интеллекта.
Мукополисахаридозы всех типов, кроме синдрома Гунтера, наследуются по аутосомно-рецессивному типу. При синдроме Гунтера тип наследования рецессивный, сцепленный с полом, нередки семейные случаи заболевания.
Мукополисахаридоз VII типа обусловлен дефектом р-глюкуронидазы и по клиническим проявлениям напоминает мукополисахаридоз I типа, но без помутнения роговицы; тип наследования неизвестен.
Наиболее типичным симптомом поражения глаз при мукополисахаридозах является дистрофия роговицы. Наиболее рано помутнение роговицы развивается при синдроме Марото — Лами. Большинство исследователей не обнаруживают изменений глаз при синдроме Санфилиппо. Синдром Моркио также протекает с дистрофией роговицы, но она нередко появляется поздно, чаще у детей старше 10 лет. Дистрофию роговицы обнаруживают у 75— 90% больных мукополисахаридозами.
Из других симптомов при мукополисахаридозах описаны мегалокорнеа, гидрофтальм, атрофия и застой соска зрительного нерва, а также дегенерация сетчатки и глаукома.
При мукополисахаридозах проводят полное исследование глаза и его функций. Неврологические и генетические исследования выполняют по классической схеме.

Дифференциальную диагностику форм заболевания следует проводить на основе тяжести поражения органа зрения и соматических проявлений (табл. 31). У обследованных больных были выявлены следующие характерные общеклинические симптомы: увеличение и изменение формы головы, костно-суставные деформации.
Необходимо особо отметить, что мукополисахаридозы в начальных стадиях необходимо дифференцировать от рахита, гипотиреоза, инфекционного неспецифического полиартрита, полигландулярной недостаточности, липоидоза. Следует подчеркнуть также, что эти дети часто болеют простудными заболеваниями.
В дальнейшем у них выявляются грубые черты лица, и все больные отстают в росте. У них отмечаются пупочные и паховые грыжи, гепатоспленомегалия, пороки сердца. При всех формах мукополисахаридозов ранними признаками являются изменения глаз различной степени выраженности.
В процессе наблюдения за больными детьми выявлено, что наиболее ранние (у новорожденных и в первые месяцы после рождения) и тяжелые поражения глаз выявляются при мукополисахаридозе I типа. Они наблюдаются у всех больных.
У детей уже в первые месяцы жизни отмечаются длинные и густые ресницы, латеральный проптоз, пастозносгь век, особенно нижних. Конъюнктива век и глазного яблока отечная, цианотичная.
Стенки сосудов утолщены. У всех больных выявляются макрокорнеа (12—13,5 мм) без расширения лимба и дистрофия роговицы различной степени выраженности. Помутнение роговицы, от нежного облаковидного до грубого диффузного, захватывает глубокие слои роговицы и более интенсивно в лимбальной зоне.
Строма роговицы всегда утолщена. Может наблюдаться застойный сосок зрительного нерва, а затем и его атрофия. Сетчатка и особенно парапапиллярная зона отечны, сосуды глазного дна расширены, стенки их утолщены. Переднезадняя ось глазного яблока увеличена у всех больных по сравнению с нормой на 2 -2,5 мм. Ни у одного больного не было выявлено повышения внутриглазного давления.
Синдром Гунтера выявляется преимущественно у мальчиков в возрасте 3 лет и старше. Ранние изменения скелета в виде увеличения размера черепа расценивают как признаки рахита. Тугоподвижность и изменения формы суставов становятся заметными к 3 годам, а иногда в более старшем возрасте. Изменения скелета в виде деформации грудной клетки и позвоночника выражены умеренно.
Изменения органа зрения при синдроме Гунтера характеризуются ранним увеличением роговицы и глазного яблока, но менее выраженным, чем при синдроме Гурлер. Помутнение роговицы выявляется к 4 - 6 годам, а иногда и позже. При биомикроскопическом исследовании выявляют, что наиболее интенсивное помутнение отмечается на 3 и 9 часах у лимба в глубоких слоях роговицы, строма которой утолщена.
В строме радужки, преимущественно на 3 и 9 часах, определяются новообразованные кровеносные сосуды. На глазном дне при офтальмоскопии обнаруживают расширенные кровеносные сосуды с утолщенной стенкой, а также застойные соски или частичную атрофию дисков зрительных нервов. Глазные симптомы предшествуют изменениям скелета и внутренних органов.
Синдром Санфилиппо характеризуется в первую очередь снижением интеллекта. Отмечается слабовыраженная гепатоспленомегалия. Отставания в росте не замечено. Имеются тугоподвижность суставов и изменение их формы.
Из глазных симптомов при синдроме Санфилиппо наблюдаются: гипертелоризм и небольшой латеральный проптоз. Роговица прозрачна, размер ее в норме. На глазном дне выявляют некоторое расширение калибра вен, т. е. картина синдрома отличается от таковой при мукополисахаридозах I и II типов.
Синдром Моркио отличается значительными деформациями скелета.
Синдром Шейе внешне имеет много общего с инфекционным неспецифическим полиартритом. Основными характерными симптомами заболевания являются изменения формы мелких и средних суставов, их болезненность и тугоподвижность.
Изменения глаз при синдроме Шейе характеризуются признаками врожденной глаукомы, возникающими уже в ранних стадиях заболевания. Позднее, когда присоединяются боли и происходит деформация мелких суставов, появляются такие же изменения глаз, как при коллагенозном увейте, с выраженной дистрофией роговицы, которая сопровождается буллезным перерождением. В начальной стадии помутнение локализуется в зоне передней пограничной пластинки, причем более интенсивное оно у лимба, а затем распространяется на все слои роговицы.
Наличие у больных тотального помутнения увеличенной роговицы с утолщенной стромой на фоне выраженного отека конъюнктивы свидетельствует о мукополисахаридозе. Диагноз подтверждают результаты определения содержания КГАГ в моче больных. Необходимо отметить, что наряду с резко выраженными клиническими формами заболевания встречаются стертые формы мукополисахаридозов, при которых ведущими признаками являются только поражения глаз.
В заключение необходимо указать, что результаты офтальмологических исследований в сочетании с общими симптомами заболевания позволяют диагностировать мукополисахаридоз и уточнить его тип. При определении экскреции КГАГ с мочой у больных мукополисахаридозом выявляют их увеличение в десятки раз по сравнению с нормой.
В то же время суточная экскреция оксипролина у всех этих больных оказывается сниженной. Особенно высокое содержание КГАГ в моче наблюдается у больных с синдромом Гурлер. Биохимическое изучение фракционного состава КГАГ показывает, что наряду с различиями имеется много общего в качественном составе КГАГ при всех типах мукополисахаридозов. У больных с синдромом Гурлер и Гунтера увеличено также выделение гепарансульфата. Особенно высокая гепарансульфатурия наблюдается при синдроме Санфилиппо.
Кроме описанных синдромов, характеризующихся поражением соединительной ткани, в офтальмологической, педиатрической и неврологической практике особое место занимают синдромы Марфана, Маркезани, гомоцистинурии и др.
Синдром Марфана (долихостеномиелия) - врожденное заболевание, характерными признаками которого являются высокий рост, необычная длина конечностей, тонкие длинные паукообразные пальцы, слабое развитие мышечной ткани. В последующие годы заболевание было названо арахнодактилией.
Тип наследования синдрома Марфана аутосомно-доминантный. Характерным симптомом заболевания является эктопия хрусталиков. Описаны пороки развития и других органов при синдроме Марфана, однако большинство исследователей считают наиболее типичными признаками заболевания поражение глаз, деформацию скелета и нарушения деятельности сердечно-сосудистой системы.
Из поражений глаз, кроме эктопии хрусталика, наблюдаются мегалокорнеа, кератоконус, голубые склеры, высокая близорукость, отслойка сетчатки. Нередко при синдроме Марфана развивается глаукома.
Для объективизации диагноза синдрома Марфана применяют исследование КГАГ. При синдроме Марфана экскреция КГАГ с мочой увеличена в 2—3 раза по сравнению с нормой.
Наиболее ранним глазным проявлением синдрома Марфана можно считать гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы.

Из других глазных симптомов встречаются эпикантус, эмбриотоксон, кератоконус, косоглазие, катаракта, микро- и сферофакия, врожденная глаукома (табл. 32). Изменения на глазном дне отмечаются у детей старшего возраста.
Миопическая рефракция, о наличии которой свидетельствуют результаты эхобиометрии толщины хрусталика (на 1—2 мм больше по сравнению с нормой), обусловлена более высокой преломляющей способностью хрусталика вследствие изменения его сферичности под влиянием разрыва волокон ресничного пояска. Сагиттальная ось глаза нормальная. Не исключено развитие и усиление близорукости вследствие образования кератоконуса. Иначе говоря, имеются основания утверждать, что близорукость при синдроме Марфана имеет чисто рефракционную, но не осевую природу.
Результаты биохимических исследований свидетельствуют о том, что изменения органа зрения при синдроме Марфана связаны в основном с увеличением содержания коллагена, о чем свидетельствует оксинупролинурия. Увеличение экскреции КГАГ, особенно гиалуроновой кислоты, отягощает течение основного заболевания.
В клинико-офтальмологических исследованиях установлено, что очень сходная картина глазных изменений обнаруживается и при других заболеваниях, в частности при гомоцистинурии (табл. 33).

Эктопия в сочетании с другими симптомами были выявлены у больных с синдромом Маркезани. Эти больные маленького роста, череп у них имеет брахицефалическую форму, туловище, шея и конечности короткие. Мышцы и подкожная жировая клетчатка хорошо выражены. Тип наследования доминантный и аутосомно-рецессивный.
Поражения глаз при синдроме Маркезани характеризуются тем, что передняя камера в центре мелкая, а на периферии глубокая, а также дрожанием радужки по периферии. Отдельные участки радужки атрофичны.
Редко наблюдается аниридия. Хрусталик имеет шаровидную форму, уменьшен в размере. Хорошо видны волокна ресничного пояска. В некоторых случаях хрусталик смещен книзу. Наблюдаются случаи вывиха хрусталика в переднюю камеру или стекловидное тело.
Глазные изменения в виде истончения и повышенной прозрачности склер наблюдаются при синдроме В а н-д е р-Х е в е. Для заболевания характерна тугоухость. Синдром голубых склер относится к конституциональным особенностям соединительной ткани, обусловленным множественными генными дефектами; наследуется по аутосомно-доминантному типу.
Заболевание связано с изменением эластических и коллагеновых волокон. У 60% больных склеры имеют голубую окраску. Причиной изменения окраски склер является тонкая наружная оболочка глаза, через которую просвечивается сосудистая оболочка.
Серовато-синяя окраска склер наблюдается уже у новорожденных. Наблюдающаяся в норме у новорожденных голубоватая окраска склер к 6 мес постепенно, исчезает, и у здоровых детей они становятся белесоватыми.
У больных наблюдаются утолщение мелких суставов конечностей и позвоночника, их деформация и ограничение подвижности. Выражен остеопороз. Отмечаются задержка закрытия родничка и развития зубов, черты лица, характерные для больного гаргоилизмом, большой язык, кифосколиоз, изменения ЭЭГ.
Умственное развитие детей не страдает. Клиническая картина сходна с проявлениями мукополисахаридоза. Однако содержание КГАГ в моче у таких больных в норме. При гистохимическом исследовании в коже выявляются высокое содержание уроновой кислоты и метахромазия культуры фибробластов.
Прогноз в отношении зрения малоблагоприятный, но отмечен эффект от применения больших доз ретинола (витамина А).
Таким образом, глазные поражения при болезнях соединительной ткани характеризуются полиморфизмом.
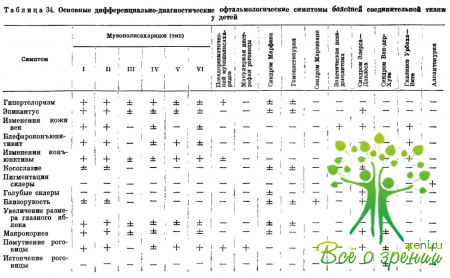
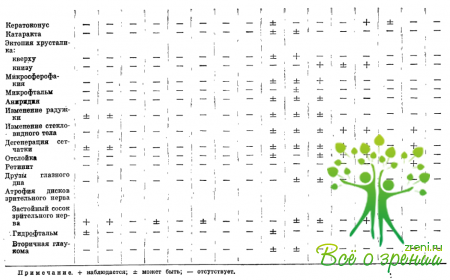
Изучение ранних офтальмологических симптомов позволяет дифференцировать отдельные формы заболеваний (табл. 34). Офтальмологические исследования помогают понять важные стороны патогенеза изучаемых заболеваний, уточнить роль изменений коллагена и основного вещества соединительной ткани в их развитии.
Изучение обмена КГАГ и оксипролина позволяет уточнить механизмы развития офтальмологических симптомов при болезнях соединительной ткани, а следовательно, принять соответствующие меры для предупреждения их возникновения и лечения.
Читайте также: