Болезнь Крейтцфельдта-Якоба
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
Болезнь Крейтцфельда–Якоба (БКЯ) является редким нейродегенеративным заболеванием, обусловленным накоплением патологической изоформы прионного белка. Классическая клиническая картина БКЯ характеризуется быстро прогрессирующей деменцией, атаксией, миоклонией, а также акинетическим мутизмом в терминальной стадии заболевания. Из инструментальных методов исследования ведущая роль в клинической практике принадлежит магнитно-резонансной томографии головного мозга. С 2014 по 2019 г. в Республике Саха (Якутия) авторы наблюдали 4 пациентов с вероятной БКЯ. У всех пациентов были примерно одинаковые возраст дебюта заболевания (50–60 лет) и начало с неспецифических церебральных симптомов. Однако последующее развитие быстро прогрессирующей деменции и других характерных признаков позволило предположить БКЯ. У пациентов выявлены показательные нейровизуализационные признаки в виде гиперинтенсивности хвостатых ядер и подушек таламуса в режимах FLAIR и DWI с образованием типичного сигнала «хоккейных клюшек», а также гиперинтенсивность серого вещества в режиме DWI (симптом «ожерелья Венеры»). У 3 пациентов заболевание закончилось фатально в течение года после его дебюта. Четвертая пациентка находится под домашним наблюдением (длительность заболевания 6 мес).
Авторы полагают, что в настоящее время уровень диагностики БКЯ недостаточен из-за схожести ее клинических симптомов в дебюте с другими расстройствами, включая цереброваскулярные и нейродегенеративные заболевания.
Ключевые слова
Об авторах
ФГБНУ «Якутский научный центр комплексных медицинских проблем»
Россия
Россия, 677000, Якутск, Сергеляхское шоссе, 4
ФГБНУ «Якутский научный центр комплексных медицинских проблем»; ФГАОУ ВО «Северо-Восточный федеральный университет им. М.К. Аммосова»
Россия
Россия, 677000, Якутск, Сергеляхское шоссе, 4
Россия, 677000, Якутск, ул. Белинского, 58
ФГБНУ «Якутский научный центр комплексных медицинских проблем»
Россия
Россия, 677000, Якутск, Сергеляхское шоссе, 4
M.K. Ammosov North-Eastern Federal University
Россия
Россия, 677000, Якутск, ул. Белинского, 58
ФГБНУ «Якутский научный центр комплексных медицинских проблем»
Россия
Россия, 677000, Якутск, Сергеляхское шоссе, 4
ФГБНУ «Якутский научный центр комплексных медицинских проблем»
Россия
Россия, 677000, Якутск, Сергеляхское шоссе, 4
ГБУ Республики Саха (Якутия) «Республиканская больница №2 – Центр экстренной медицинской помощи»
Россия
Россия, 677000, Якутск, ул. Петра Алексеева, 83А
Список литературы
1. Шнайдер НА. Болезнь КрейцфельдаЯкоба: новый взгляд на старую проблему (история изучения, этиология и патогенез). Журнал неврологии и психиатрии им. С.С. Корсакова. 2013;113(4):72-9.
2. Jacobs DA, Lesser RL, Mourelatos Z, et al. The Heidenhain variant of Creutzfeldt-Jakob disease: clinical, pathologic, and neuroimaging findings. J Neuroophthalmol. 2001;21(2):99-102. doi: 10.1097/00041327-200106000-00008.
3. Uttley L, Carroll C, Wong R, et al. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020;20(1):e2-e10. doi: 10.1016/S1473-3099(19)30615-2.
5. Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006 Jan 24;66(2):286-7. doi: 10.1212/01.wnl.0000196440.00297.67.
6. Manix M, Kalakoti P, Henri M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2. doi: 10.3171/2015.8.FOCUS15328.
7. Parchi P, De Boni L, Saverioni D, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012 Oct;124(4):517-29. doi: 10.1007/s00401-012-1002-8.
8. Fragoso DC, Goncalves Filho AL, Pacheco FT, et al. Imaging of Creutzfeldt-Jakob disease: imaging patterns and their differential diagnosis. Radiographics. 2017;37(1):234-257. doi: 10.1148/rg.2017160075.
9. Hirst CL. Sporadic Creutzfeldt-Jakob disease presenting as a stroke mimic590. Br J Hosp Med (Lond). 2011;72(10):590-591. doi: 10.12968/hmed.2011.72.10.590.
10. Hanumanthu R, Alchaki A, Nyaboga A, et al. An unusual case of sporadic CreutzfeldJacob disease presenting as acute neuropathy. Mov Disord. 2017;32:563-4.
12. Pachalska M, Kurzbauer H, ForminskaKapuscik M, et al. Atypical features of dementia in a patient with Creutzfeldt-Jakob disease. Med Sci Monit. 2007 Jan;13(1):CS9-19. Epub 2006 Dec 18.
13. Litzroth A, Cras P, De Vil B, Quoilin S. Overview and evaluation of 15 years of Creutzfeldt-Jakob disease surveillance in Belgium, 1998–2012. BMC Neurol. 2015 Dec 2;15:250. doi: 10.1186/s12883-015-0507-x.
14. Mahboob HB, Kaokaf KH, Gonda JM. Creutzfeldt-Jakob Disease Presenting as Expressive Aphasia and Nonconvulsive Status Epilepticus. Case Rep Crit Care. 2018:5053175. doi: 10.1155/2018/5053175.
15. Ali R, Baborie A, Larner AJ, et al. Psychiatric presentation of sporadic Creutzfeldt-Jakob disease: a challenge to current diagnostic criteria. J Neuropsychiatry Clin Neurosci. 2013;25(4):335-8. doi: 10.1176/appi.neuropsych.13020025.
16. Rodriguez-Porcel F, Ciarlariello VB, Dwivedi AK, et al. Movement Disorders in Prionopathies: A Systematic Review. Tremor Other Hyperkinet Mov (NY). 2019;9. doi: 10.7916/tohm.v0.712. eCollection 2019.
17. Sakuma T, Watanabe S, Ouchi A, et al. Three Cases of Creutzfeldt-Jakob Disease with Visual Disturbances as Initial Manifestation. Case Rep Ophthalmol. 2019 Oct 23;10(3): 349-356. doi: 10.1159/000503274. eCollection 2019 Sep-Dec.
19. Thompson A, MacKay A, Rudge P, et al. Behavioral and psychiatric symptoms in prion disease. Am J Psychiatry. 2014;171(3):265-274. doi: 10.1176/appi.ajp.2013.12111460.
20. Переседова АВ, Стойда НИ, Гнездицкий ВВ и др. Спорадическая болезнь Крейтцфельдта-Якоба: клиническое наблюдение. Анналы клинической и экспериментальной неврологии. 2011;5(4):52-6.
22. Vitali P, Maccagnano E, Caverzasi E, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76(20):1711-1719. doi: 10.1212/WNL.0b013e31821a4439.
23. Zerr I, Kallenberg K, Summers D, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(10): 2659-2668. doi: 10.1093/brain/awp191.
24. Geschwind MD. Prion diseases. Continuum (Minneap Minn). 2015 Dec;21(6 Neuroinfectious Disease):1612-38. doi: 10.1212/CON.0000000000000251.
26. Satoh K, Fuse T, Nonaka T, et al. Postmortem Quantitative Analysis of Prion Seeding Activity in the Digestive System. Molecules. 2019 Dec 16;24(24). pii: E4601. doi: 10.3390/molecules24244601.
28. Klug GM, Wand H, Boyd A, et al. Enhanced geographically restricted surveillance simulates sporadic Creutzfeldt-Jakob disease cluster. Brain. 2009 Feb;132(Pt 2):493-501. doi: 10.1093/brain/awn303. Epub 2008 Nov 28.
29. Klug GM, Wand H, Simpson M, et al. Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol Neurosurg Psychiatry. 2013 Dec;84(12):1372-7. doi: 10.1136/jnnp-2012-304820. Epub 2013 Aug 21.
Болезнь Крейтцфельдта-Якоба (БКЯ)
Болезнь Крейтцфельда–Якоба (БКЯ) является наиболее распространенной человеческой прионной болезнью. Она наблюдается во всем мире и имеет несколько форм и подвидов. Симптомы БКЯ включают в себя слабоумие, миоклонус и другие патологии центральной нервной системы; смерть обычно происходит между 4 месяцами ми и 2 годами после возникновения, в зависимости от формы и подтипа БКЯ. Лечение носит поддерживающий характер.
сБКЯ является наиболее распространенным типом, на который приходится около 85% случаев. сБКЯ обычно проявляется у пациентов в возрасте > 40 лет (медиана – около 65 лет).
Семейная БКЯ диагностируется в около 5 до 15% случаев. Наследование аутосомно-доминантное, возраст начала заболевания, как правило, раньше, чем при сБКЯ с большей длительностью заболевания.
Вариантная БКЯ (вБКЯ)
ВБКЯ редкая приобретенная форма БКЯ. Большинство случаев произошло в Великобритании (UK) - 178 случаев по состоянию на 3 февраля 2020 года по сравнению с 54 случаями во всех других европейских и неевропейских странах по состоянию на декабрь 2019 года. вБКЯ диагностировалась после приема в пищу мяса, полученного от крупного рогатого скота, заболевшего губкообразной энцефалопатией крупного рогатого скота (ГЭКРС), также называемой коровьим бешенством.
В случае заражения вБКЯ, симптомы развиваются в более раннем среднем возрасте ( 30 лет), чем в случае заражения сВКЯ. В недавно диагностированных случаях инкубационный период (время между употреблением в пищу зараженной говядины и развития симптомов) составлял от 12 до более чем 20 лет.
В начале 1980-х годов из-за слабо контролируемых правил переработки побочных продуктов животного происхождения, зараженной ткани, вероятно, от овец, зараженных скрепи, или крупного рогатого скота, зараженных ГЭКРС, скрепи прионового белка (PrP Sc ) попадали в корм для крупного рогатого скота. У сотен тысяч голов крупного рогатого скота развилась ГЭКРС. Несмотря на широкое воздействия, относительно у немногих людей, которые употребляли в пищу мясо больного крупного рогатого скота развилась вБКЯ.
В связи с длительным инкубационным периодом ГЭКРС связь между заболеванием и зараженным мясом в Великобритании не была установлена до тех пор, пока заболеваемость ГЭКРС не переросла в эпидемию. Эпидемия ГЭКРС перешла под контроль после массивного убоя скота и после изменения в процедурах производства технических фабрикатов, которые резко сократили загрязнения мяса тканями нервной системы. В Великобритании ежегодное число новых случаев вБКЯ, которое достигло пика в 2000 году, неуклонно сокращалось, и было зафиксировано только 2 случая после 2011 года.
Четыре случая вБКЯ были связаны с переливанием крови, они диагностировались у людей, которым была проведена трансфузия между 1996 и 1999. В Великобритании приблизительно 1 из 2000 человек могут быть носителями вБКЯ (на основании изучения большого количества образцов ткани аппендикса), но не имеют никаких симптомов; эти люди могут передавать болезнь, являясь донорами крови или при проведении хирургической процедуры. Таким образом, неясно, имеется ли группа пациентов, которым переливали инфицированную кровь и которые, таким образом, находятся в зоне риска последующего развития вБКЯ. Тем не менее, новые направления в критериях, связанных с донорством, связанные с вБКЯ, могут дополнительно снизить риск передачи вБКЯ при переливании крови, который уже достаточно низкий за пределами Франции и Великобритании.
Общие справочные материалы
Симптомы и признаки БКЯ
Приблизительно у 70% пациентов с болезнью Крейтцфельда-Якоба имеются нарушения памяти, снижение внимания и изменение поведения, которые в конечном счете развиваются у всех пациентов; у 15–20% отмечается расстройство координации и атаксия, которые часто появляются на ранних стадиях заболевания. На более поздних стадиях могут возникнуть миоклонические судороги Миоклония Миоклония – это кратковременное быстрое сокращение мышцы или группы мышц. Диагноз устанавливают на основании клинических проявлений и иногда подтверждают результатами электромиографического. Прочитайте дополнительные сведения , вызываемые громким звуком (миоклония при испуге) или другими сенсорными стимулами. У людей с вБКЯ диагностируются психические симптомы (например, тревога, депрессия), а не потеря памяти. Более поздние симптомы похожи при обеих формах.
Помимо наиболее характерных для БКЯ деменции, атаксии и миоклонических судорог могут появиться и другие неврологические расстройства (например, галлюцинации, эпилептиформные припадки, нейропатия, различные двигательные нарушения).
При сБКЯ часто встречаются зрительные нарушения (например, дефекты поля зрения, диплопия, затуманенность или нечеткость зрения, зрительная агнозия).
Болезнь Крейтцфельдта-Якоба
Заболевания
Болезнь Крейтцфельдта–Якоба
Это быстро прогрессирующее фатальное нейродегенеративное заболевание, ключевым патогенетическим звеном которого является гибель нейронов, индуцированная прионовыми белками. Заболевание приводит к быстро прогрессирующему слабоумию и смерти обычно в течение года от начала.
История
Заболевание было названо в честь Ханса Герхарда Кройцфельдта (1885-1964), немецкого невролога, который впервые описал это состояние в 1920 году, и Альфонса Марии Якоба (1884-1931), немецкого невролога, которая также описала это состояние в отдельном исследовании в 1921 году.
Эпидемиология
Описаны три основных типа болезни Крейтцфельдта-Якоба:
- Спорадический – составляет 85-90% случаев и разделена на множество подтипов в зависимости от мутации;
- Семейный – 10% случаев, вызвана мутацией PRPc;
- Ятрогенный;
Особенности клиники
К основным клиническим проявлениям заболевания относятся: быстро прогрессирующие когнитивные нарушения, миоклонус, дистония, акинетикоригидный синдром, спастичность, гиперрефлексия, атаксия, зрительные расстройства, на поздних этапах заболевания акинетический мутизм. Примерно в трети случаев отмечаются эпилептические припадки. Средняя продолжительность жизни при спорадической форме БКЯ составляет около 5 мес; более 90% пациентов умирают в течение 1 года из-за аспирационной пневмонии в состоянии акинетического мутизма. Используемые в настоящее время диагностические критерии БКЯ включают быстро прогрессирующую деменцию, экстрапирамидно/ пирамидные и зрительные расстройства, миоклонус, мозжечковую атаксию, а также характерные изменения ЭЭГ в виде комплексов высокоамплитудных 2–3-фазных острых волн и положительный маркер белок 14-3-3 в цереброспинальной жидкости (ЦСЖ). Сложности диагностики БКЯ связывают с редкостью заболевания, клиническим полиморфизмом и недостаточной информированностью врачей.
До недавнего времени «золотым стандартом» верификации диагноза БКЯ являлась биопсия головного мозга, позволяющая выявить характерные изменения в мозговой ткани в виде мелких вакуолей в телах нейронов, из-за чего ткань мозга приобретает губчатый вид, пролиферации клеток глии при отсутствии признаков воспаления. При электронной микроскопии возможно обнаружение прионных палочек, являющихся патогномоничным признаком заболевания. Указанные морфологические изменения отмечаются в коре головного мозга, базальных ганглиях, мозжечке и верхних отделах ствола мозга. Однако в случаях БКЯ биопсия мозга не нашла широкого применения в клинической практике из-за инвазивности метода, сложности санитарной обработки оборудования и утилизации биоматериалов, связанных с высокой устойчивостью прионов, а также вследствие небольшого объема биоптата мозговой ткани, что может быть причиной ложноотрицательных результатов морфологического и иммуногистохимического исследования. Однако в настоящее все больше распространена МРТ диагностика данного заболевания, по причине выявления специфических изменений, наиболее информативными являются последовательность DWI.
МРТ при болезни Крейтцфельдта-Якоба
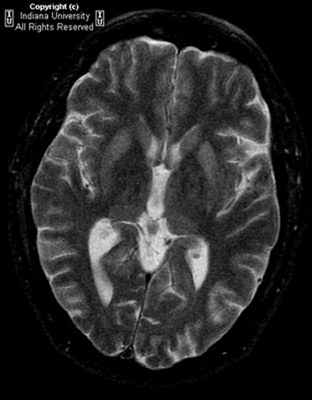
Болезнь Крейтцфельдта-Якоба – редкое дегенеративное заболевание центральной нервной системы, при котором происходит поражение коры и базальных ганглиев головного мозга, а также нарушение функций спинного мозга.
Болезнь Крейтцфельдта-Якоба (она же - губчатая энцефалопатия) встречается довольно редко и может длительное время протекать бессимптомно. Длительность инкубационного периода зависит от способа заражения: так, с момента заболевания до появления первых симптомов может пройти от 12 месяцев до 10-15 лет. Источником болезни, по распространенному мнению, считается коровье мясо, зараженное прионами*. Однако болезнь может носить наследственный характер или может быть занесена во время операции нестерильными медицинскими инструментами.
Особенности развития болезни Крейтцфельдта-Якоба
Как правило, болезнь включает три стадии развитии: продромальный период, инициальный и развернутый. Первая стадия (продромальная) характеризуется быстрой утомляемостью пациента, расстройством сна, отсутствием аппетита и прочими признаками, которые могут быть приняты, например, за проявление депрессии. Через несколько недель или месяцев наступает второй этап (инициальный), когда пациент испытывает головокружения и головные боли, ощущает ухудшение зрения.
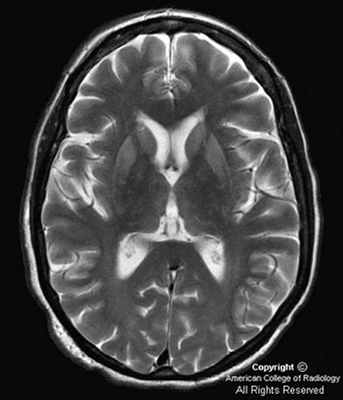
Третья стадия, развернутая, может привести к спастическому параличу и таким формам сокращения мышц, как миоклония, тремор, ригидность. На этом этапе у пациента происходит атрофия нейронов головного мозга. Кроме того, пациент постоянно чувствует напряженность в мышцах. При некоторых клинических формах болезнь на третьей стадии сопровождается дисфункцией мозжечка.
Для достоверной диагностики данного заболевания наиболее эффективным методом является прижизненная биопсия с забором вещества мозга. Однако, как правило, биопсия назначается как крайний метод диагностики. МРТ в данном случае позволяет осуществить дифференциальную диагностику, что поможет специалисту исключить большинство различных патологий.
Болезнь Крейтцфельдта-Якоба: МРТ-диагностика
Для диагностирования дегенеративного заболевания лечащий врач может назначить комплекс взаимоуточняющих методик: ЭЭГ и МРТ головного мозга.
Магнитно-резонансная томография позволяет получить качественные и контрастные изображения головного мозга пациента. Во время проведения МРТ выявляются участки повышенного сигнала, локализованные в подкорковых ганглиях и таламусах. Кроме того, магнитно-резонансная томография дает возможность успешно определять признаки атрофических изменений мозжечка, коры головного мозга и расширение желудочковой системы.
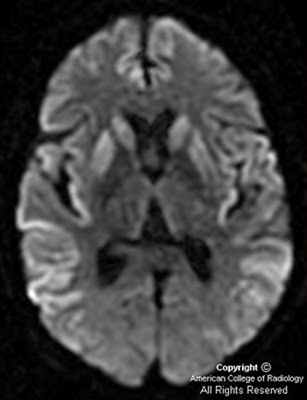
МРТ является абсолютно безвредным и безболезненным методом диагностики. МРТ является абсолютно безвредным и безболезненным методом диагностики. Высокое контрастное разрешение позволяет распознавать любую патологию структур головного мозга, оценить состояние белого и серого вещества, черепно-мозговых нервов, ствола мозга, образований задней черепной ямки.
Магнитно-резонансная диагностика часто назначается в целях дифференциальной диагностики заболеваний нервной системы. Как мы уже отметили, болезнь Крейтцфельдта-Якоба встречается довольно редко и трудно идентифицируется, поэтому при постановке такого диагноза необходимо исключить другие заболевания головного мозга. Ими могут быть лобно-височная и мультинфарктная деменция, энцефалит, хронический менингит, нормотензивная гидроцефалия, энцефалопатия и другие заболевания, некоторые из которых, имеющие схожую с болезнью Крейтцфельдта-Якоба клиническую картину, вызывают деменцию. У пациентов с расстройством интеллекта (слабоумием) МРТ также выявляет структурные изменения в головном мозге и его аномалии.
Таким образом, при диагностике болезни Крейтцфельдта-Якоба важным преимуществом МРТ (в сравнении с другими методами лучевой диагностики) является возможность исключить большинство различных патологий.
Диагностика синдрома Крейтцфельдта-Якоба

Как диагностировать синдром Крейтцфельдта-Якоба: Синдром Крейтцфельдта-Якоба (БКЯ) вызывается аномальным инфекционным белком в головном мозге, который называется прионом. Белки — это молекулы, состоящие из аминокислот, которые помогают клеткам тела функционировать. Они начинаются с цепочки аминокислот, которые затем складываются в трехмерную форму - процесс «сворачивание белков». Нормальные прионные белки обнаруживаются почти во всех тканях организма, но их самые высокие уровни находятся в мозге и нервных клетках. Точная роль нормальных прионных белков неизвестна, но считается, что они могут играть роль в передаче сигналов между определенными клетками мозга. Во время сворачивания цепочек белка иногда возникают ошибки, и неправильно свернутые прионные белки перерабатываются организмом, но они могут накапливаться в головном мозге, если не переработаются.
Прионы представляют собой неправильно свернутые прионные белки, которые накапливаются в головном мозге и вызывают неправильную укладку других прионных белков. Это заставляет клетки мозга умирать, высвобождая больше прионов для заражения других клеток. В конце концов, скопления клеток мозга погибают, и в мозгу могут появиться отложения неправильно свернутого прионного белка, называемые бляшками. Прионные инфекции также вызывают образование небольших отверстий в мозговых тканях, поэтому они становятся похожими на губку. Данные повреждения мозга вызывают психические и физические нарушения и в конечном итоге приводят к смерти пациента.
Первичная диагностика синдрома БКЯ потребует консультации невролога. В качестве дополнительного обследования по результатам осмотра врач может назначить:
- ;
- ЭЭГ;
- поясничная биопсия спинного мозга;
- биопсия миндалин мозга;
- генетический тест.
Какой врач лечит синдром Крейтцфельдта-Якоба: При симптомах данной патологии следует обратиться к врачу-неврологу.
Виды синдрома Крейтцфельдта-Якоба
Все различные типы болезни Крейтцфельдта-Якоба вызываются накоплением прионов в головном мозге. Но причина, по которой это происходит, у каждого типа разная.
Спорадический синдром Крейтцфельдта-Якоба
Несмотря на то, что случаи спорадического синдрома Крейтцфельдта-Якоба очень редки, это наиболее распространенный тип БКЯ, составляющий около 8 из 10 случаев данного заболевания. Ученым пока неизвестно, что вызывает спорадические болезни Крейтцфельдта-Якоба, но есть предположение, что нормальный прионный белок спонтанно превращается в прион, или нормальный ген спонтанно превращается в дефектный ген, который производит прионы. Спорадическая болезнь Крейтцфельдта-Якоба чаще встречается у людей, у которых есть определенные версии гена прионного белка. В настоящее время не выявлено никаких других факторов, которые увеличивают риск развития спорадической БКЯ.
Вариант синдрома Крейтцфельдта-Якоба
Существуют четкие доказательства того, что вариант синдрома Крейтцфельдта-Якоба (v болезни Крейтцфельдта-Якоба) вызывается тем же штаммом прионов, который развивает губчатую энцефалопатию крупного рогатого скота («коровье бешенство»). В 2000 году масштабное медицинское исследование показало, что прион передавался через крупный рогатый скот, который получал мясокостную смесь со следами инфицированного мозга или спинного мозга. Затем прион попадал в переработанные мясные продукты и становился частью пищевой цепочки человека. Все случаи v болезни Крейтцфельдта-Якоба произошли у пациентов с определенной версией (MM) гена прионного белка, который влияет на то, как организм вырабатывает ряд аминокислот. Некоторые эксперты считают, что, если усилить меры по контролю за продуктами питания, то количество случаев БКЯ будет снижаться. Вариант синдрома Крейтцфельдта-Якоба также может передаваться при переливании крови.
Семейный или наследственный синдром Крейтцфельдта-Якоба
Семейный или наследственный синдром Крейтцфельдта-Якоба — это редкая форма БКЯ, вызванная наследственной мутацией (аномалией) в гене, который продуцирует прионный белок. Измененный ген, по-видимому, производит неправильно свернутые прионы, вызывающие болезнь Крейтцфельдта-Якоба. У каждого есть 2 копии гена прионного белка, но мутировавший ген является доминирующим. Это означает, что пациенту нужно унаследовать только 1 мутировавший ген, чтобы развить заболевание. Таким образом, если у 1 родителя есть мутировавший ген, есть 50% -ная вероятность, что он будет передан их детям. Поскольку симптомы семейной болезни Крейтцфельдта-Якоба обычно не проявляются, пока человеку не исполнится 50 лет, многие люди с этим заболеванием не знают, что их дети также находятся в группе риска унаследовать эту патологию, когда они решат создать семью.
Ятрогенный синдром Крейтцфельдта-Якоба
Ятрогенный синдром Крейтцфельдта-Якоба — это когда инфекция передается от человека с БКЯ в результате медицинского или хирургического лечения. Большинство случаев ятрогенной болезни Крейтцфельдта-Якоба произошло из-за использования гормона роста человека для лечения детей с задержкой роста. Между 1958 и 1985 годами тысячи детей лечились гормоном, который в то время извлекался из гипофиза человеческих трупов. У меньшинства этих детей развилась болезнь Крейтцфельдта-Якоба, поскольку гормоны, которые они получали, были взяты из питуитарных желез, инфицированных БКЯ. Несколько других случаев ятрогенной болезни Крейтцфельдта-Якоба произошли после того, как пациенты вступили в контакт с хирургическими инструментами, которые были загрязнены прионами БКЯ. Поскольку прионы более жесткие, чем вирусы или бактерии, обычный процесс стерилизации хирургических инструментов не имел никакого эффекта.
Заразен ли синдром Крейтцфельдта-Якоба?
Теоретически болезнь Крейтцфельдта-Якоба может передаваться от пораженного пациента к другим, но только через инъекцию или имплантацию инфицированного мозга или нервной ткани. Нет никаких доказательств того, что спорадическая БКЯ передается при обычном повседневном контакте с больными, воздушно-капельным путем, кровью или половым контактом.
Симптомы болезни Крейтцфельдта-Якоба
Характер симптомов болезни Крейтцфельдта-Якоба может варьироваться в зависимости от типа заболевания. При спорадической болезни Крейтцфельдта-Якоба симптомы в основном влияют на работу нервной системы, и они быстро ухудшаются в течение нескольких месяцев. При варианте болезни Крейтцфельдта-Якоба сначала развиваются симптомы, влияющие на поведение и эмоции человека, а примерно через 4 месяца за ними следуют неврологические симптомы, которые ухудшаются в течение следующих нескольких месяцев.
Семейный синдром Крейтцфельдта-Якоба имеет такую же картину, что и спорадический, но часто для прогрессирования симптомов требуется больше времени - обычно около 2 лет, а не несколько месяцев. Характер ятрогенной болезни Крейтцфельдта-Якоба непредсказуем, так как он зависит от того, как человек подвергся воздействию инфекционного белка.
Начальные неврологические симптомы
Начальные неврологические симптомы спорадической болезни Крейтцфельдта-Якоба могут включать:
- трудности при ходьбе, вызванные проблемами равновесия и координации;
- невнятная речь;
- онемение или "булавки и иглы" в разных частях тела;
- головокружение;
- проблемы со зрением, такие как двоение в глазах и галлюцинации.
Начальные психологические симптомы
Начальные психологические симптомы варианта болезни Крейтцфельдта-Якоба могут включать:
- тяжелая депрессия;
- сильное чувство отчаяния;
- уход от семьи, друзей и окружающего мира;
- беспокойство:
- раздражительность;
- проблемы со сном (бессонница).
Расширенные неврологические симптомы
Расширенные неврологические симптомы всех форм болезни Крейтцфельдта-Якоба могут включать:
- потеря физической координации, которая может влиять на широкий спектр функций, таких как ходьба, речь и равновесие (атаксия);
- мышечные подергивания и спазмы;
- потеря контроля над мочевым пузырем и кишечником;
- слепота;
- затруднения глотания (дисфагия);
- потеря речи;
- потеря произвольного движения.
Расширенные психологические симптомы
Расширенные психологические симптомы всех форм болезни Крейтцфельдта-Якоба включают:
- потеря памяти;
- проблемы с концентрацией;
- спутанность сознания;
- чувство волнения;
- агрессивное поведение;
- потеря аппетита, что может привести к потере веса;
- паранойя;
- необычные и неуместные эмоциональные реакции.
По мере того, как состояние прогрессирует до последней стадии, больные со всеми видами синдрома Крейтцфельдта-Якоба становятся полностью прикованы к постели. Часто они совершенно не осознают свое окружение и нуждаются в круглосуточном уходе. Они также часто теряют способность говорить и не могут общаться со своими опекунами. Смерть неизбежно наступит, как правило, в результате пневмонии или дыхательной недостаточности, когда легкие перестают работать, и человек не может дышать.
Диагностика синдрома Крейтцфельдта-Якоба
Диагноз болезни Крейтцфельдта-Якоба (БКЯ) обычно основывается на истории болезни, симптомах и серии тестов. На начальном этапе обследований невролог будет проводить тесты, чтобы исключить другие заболевания с похожими симптомами, такие как болезнь Альцгеймера, болезнь Паркинсона или опухоль мозга. Для этого пациенту назначат сделать:
- - используются сильные магнитные поля и радиоволны, чтобы произвести детальное изображение мозга; могут показать отклонения в мозговых тканях, связанных с синдромом Крейтцфельдта-Якоба;
- ЭЭГ - регистрирует активность мозга и может подобрать аномальные электрические модели, встречающиеся в спорадической болезни Крейтцфельдта-Якоба;
- поясничная биопсия спинного мозга - это процедура, при которой игла вставляется в нижнюю часть позвоночника, чтобы взять образец спинномозговой жидкости и проверить биоматериал на наличие определенного белка, характерного для болезни Крейтцфельдта-Якоба;
- биопсия миндалин мозга для оценки наличия аномальных прионов;
- генетический тест, чтобы узнать, есть ли у пациента мутация в гене, вырабатывающем нормальный белок.
Положительный результат может указывать на семейное (наследственное) прионное заболевание Единственный способ подтвердить диагноз болезни Крейтцфельдта-Якоба - исследовать ткань головного мозга путем проведения биопсии. Во время биопсии головного мозга хирург просверливает крошечное отверстие в черепе и удаляет небольшой кусочек ткани мозга с помощью очень тонкой иглы. Поскольку биопсия головного мозга сопряжена с риском повреждения мозга или судорог, она выполняется только в нескольких случаях, когда есть опасения, что у пациента нет болезни Крейтцфельдта-Якоба, а есть другое заболевание, которое поддается лечению.
Лечение
В настоящее время лечение синдрома Крейтцфельдта-Якоба заключается в том, чтобы помочь больному чувствовать себя максимально комфортно и уменьшить симптомы с помощью лекарств. Например, психологические симптомы болезни, такие как тревога и депрессия, можно лечить седативными средствами и антидепрессантами, а мышечные судороги или тремор можно лечить такими лекарствами, как клоназепам и вальпроат натрия. Боль на поздних этапах заболевания можно облегчить с помощью сильнодействующих обезболивающих на основе опиатов. По мере прогрессирования синдрома Крейтцфельдта-Якоба людям с этим заболеванием потребуется серьезный уход и практическая поддержка. Некоторым людям может потребоваться помощь не только в кормлении, мытье и передвижении, но и в мочеиспускании.
Профилактика
Хотя синдром Крейтцфельдта-Якоба (БКЯ) встречается очень редко, его бывает трудно предотвратить. Это связано с тем, что большинство случаев возникают спонтанно по неизвестной причине (спорадическая болезнь Крейтцфельдта-Якоба), а некоторые вызваны наследственной генетической ошибкой (семейная болезнь Крейтцфельдта-Якоба). Методы стерилизации, используемые для предотвращения распространения бактерий и вирусов, также не полностью эффективны против инфекционного белка (приона). Но ужесточение правил повторного использования хирургического оборудования означает, что случаи распространения болезни Крейтцфельдта-Якоба через лечение (ятрогенная болезнь Крейтцфельдта-Якоба) сейчас очень редки. Также принимаются меры для предотвращения распространения варианта болезни Крейтцфельдта-Якоба через пищевую цепочку и поставку крови, используемой для переливания.
Поскольку связь между губчатой энцефалопатией крупного рогатого скота («коровье бешенство») и вариантом синдрома Крейтцфельдта-Якоба была подтверждена, был введен строгий контроль, чтобы предотвратить попадание инфицированного мяса в пищевую цепь человека. Эти элементы контроля качества мясопродуктов включают:
Читайте также: