Дефект Хагемана - диагностика, лечение
Добавил пользователь Skiper Обновлено: 28.01.2026
Данное заболевание считается достаточно редким нарушением коагуляционного гемостаза. Впервые патология была выявлена О. Ратновым в 1964 г.
Заболевание характеризуется сильным снижением активности пускового фактора внутреннего механизма свертывания крови - фактора XII.
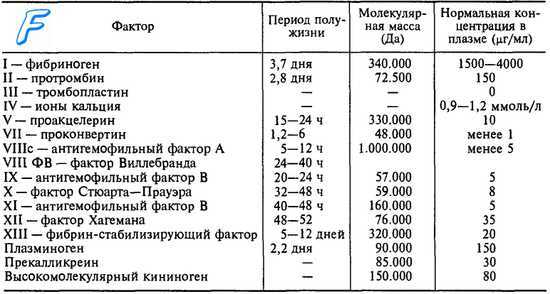
Фактор XII (фактор Хагемана) - сиалогликопротеин, активирующийся коллагеном, является естественным активатором как свертывающей, так и калликреин-кининовой и фибринолитической систем.
Что провоцирует / Причины Дефецита фрктора XII:
В большинстве случаев дефект Хагемана наследуется по рецессивно-аутосомному типу. Однако в некоторых семьях выявлено аутосомно-доминантное наследование. Видимо, синтез фактора XII контролируется двумя аутосомными генами (бимодальный тип наследования).
Еще раньше такой вывод был сделан рядом авторов на основании того, что рецессивные передатчики дефекта Хагемана подразделяются на группы с низким и более высоким содержанием фактора XII в плазме.
Иммунологические исследования свидетельствуют о том, что обе формы дефекта Хагемана (рецессивно и доминантно наследуемые) характеризуются сниженным синтезом этого белка, а не образованием его аномальных молекул.
Симптомы Дефецита фрктора XII:
При дефиците фактора XII отсутствуют какие-либо геморрагические явления (не только спонтанные, но и при травмах), несмотря на выраженное удлинение времени свертывания крови - до 30 мин и более.
Выдвинут ряд гипотез для объяснения этого парадоксального явления: отмечена способность тканевых экстрактов и тромбоцитов перекрывать дефицит фактора XII и отчасти - РТА.
Недостаточностью фибринолиза объясняется то, что у ряда больных с дефектом Хагемана, несмотря на резкое замедление свертываемости крови, отмечаются тяжелые и даже смертельные тромбоэмболические осложнения, в том числе эмболия легочной артерии после перелома тазовых костей у первого выявленного носителя этого дефекта -Хагемана.
При этом дефекте наблюдались также инфаркты миокарда, тромбофлебит у новорожденного.
Между степенью нарушения свертываемости крови и дефицитом фактора Хагемана наблюдается строгое соответствие: при резко выраженной гипокоагуляции уровень этого фактора в плазме не превышает 2% и чаще - ниже 1%; при умеренном нарушении свертываемости он варьирует от 3 до 9%.
В тех случаях, когда концентрация фактора XII в плазме составляет 10% и более, свертываемость крови, активированное парциальное тромбопластиновое время и другие тесты нормализуются.
Диагностика Дефецита фрктора XII:
Дефект Хагемана необходимо заподозрить в тех случаях, когда имеется существенное увеличение времени свертывания крови, сопровождающееся отсутствием кровоточивости или незначительной ее выраженностью. Однако наиболее полная аргументация диагноза достигается тестами смешивания с плазмой, взятой от больных с дефицитом фактора XII и других факторов, а также иммунологическим определением в плазме больного фактора XII.
Своеобразная форма дефицита фактора XII с умеренно выраженным геморрагическим синдромом (легкое появление синяков, кровотечения при операциях, травмах, иногда в родах), различными аллергическими синдромами (ангионевротический отек, бронхиальная астма, экзема) и высокой частотой ранних мозговых инсультов выявлена в 1970 г. В отличие от обычного дефекта Хагемана эта форма наследуется по неполному доминантному типу.
Лечение Дефецита фрктора XII:
Если нет полной уверенности в правильности диагноза или у больного (его родственников) раньше отмечались какие-либо кровотечения, то до хирургического вмешательства врач назначает переливание плазмы. Трансфузионная терапия проводится так же, как и при дефиците фактора XI.
При дефекте Хагемана не следует применять аминокапроновую кислоту и другие ингибиторы фибринолиза, поскольку эту патологию сопровождает недостаточность фибринолитической системы. В такой ситуации назначение антифибринолитиков повышает риск тромбоэмболии.
К каким докторам следует обращаться если у Вас Дефицит фактора XII:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Дефецита фрктора XII, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Синдром Хагемана
Дефицит фактора Хагемана (XII фактор свертывания крови) — это редкое (около 1:1 000 000 населения) и наследственно обусловленное нарушение коагуляционного гемостаза. Наследуется этот дефект преимущественно аутосомно-рецессивно, но в единичных случаях — более тяжелых — выявляется аутосомно-доминантное наследование. В иммунологических исследованиях было показано, что синдром Хагемана характеризуется сниженным синтезом фактора Хагемана, а не образованием его аномальных молекул.
В 1954 году в Кливленде (США) гематолог Оскар Ратнов наблюдал пациента Джона Хагемана 37 лет со значительно увеличенным временем кровотечения. Обратив внимание на то, что, несмотря на изменения в коагуляции, пациент перенес хирургическую операцию без значительного кровотечения, Ратнов совместно с биохимиком Эрлом Дэйви установил, какого именно белка не хватало в плазме у этого пациента, заподозрив в этом причину нарушения свертываемости крови. Этот сложный белок и был назван фактором Хагемана, а после того как английский гематолог Роберт Макфарлан в 1964 г. сформулировал каскадную теорию гемостаза, занял в ней место как 12‑й фактор свертывания.
Кровотечение — одно из самых опасных для организма состояний, защита организма от него должна быть как надежной, так и саморегулирующейся, чтобы остановка кровотечения не переходила в распространенный тромбоз. Эту роль выполняют сразу несколько систем — клеточная (тромбоцитарный гемостаз) и сложная система белков плазмы, состоящая из многих взаимосвязанных факторов свертывания:
- I — Фибриноген
- II — Протромбин
- III — Тканевой тромбопластин
- IV — Ионы кальция
- V — Проакцелерин
- VI — Акцелерин — изъят из классификации, так как является
активированным V фактором - VII — Проконвертин
- VIII — Антигемофильный фактор
- IX — Фактор Кристмаса
- X — Фактор Стюарта—Прауэра
- XI — Плазменный предшественник тромбопластина
- XII — Фактор Хагемана
- XIII — Фибринстабилизирующий фактор
- Фактор Флетчера — плазменный прекалликреин
- Фактор Фитцжеральда — высокомолекулярный кининоген
- Фактор Виллебранда — опосредует связывание тромбоцитов с субэндотелием
Фактор Хагемана — сиалогликопротеин — сложное органическое соединение, помимо белковой части включающее в себя олигосахарид и сиаловые кислоты. Сиалогликопротеин синтезируется в печени, далее свободно и «без дела» циркулирует в плазме крови, пока не соприкоснется с отрицательно заряженными поверхностями, например, коллагеном поврежденной ткани или с калликреином, о котором стоит рассказать подробнее.
Калликреин — один из основных функциональных элементов сложной калликреин-кининовой системы, регулирующей процессы воспаления, свертывания крови, микроциркуляции и функции сосудистой стенки.
Фактор Хагемана, активированный повреждением ткани, участвует в запуске калликреин-кининовой системы, превращая прекалликреин в калликреин, а калликреин в свою очередь активирует все новые молекулы фактора Хагемана.
Получается петля для каскадного нарастания процесса. За счет этого небольшое количество поврежденных тканей вызывает реакцию в значительной части плазмы крови.
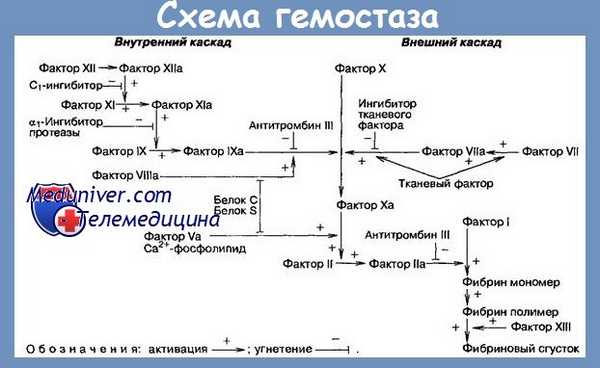
Кроме того, на 12 фактор (Хагемана) в активной форме действует 13 фактор (фибринстабилизирующий фактор), что запускает каскад биохимических реакций среди молекул, исходно присутствовавших в крови, — внутренний путь коагуляции. В отличие от внутреннего пути, внешний запускается попаданием в кровь тканевого тромбопластина из поврежденных тканей.
Таким образом, фактор Хагемана оказывается связующим звеном между процессами воспаления и свертывания крови; до его открытия связь этих процессов была гипотетической.
Клиническая картина
Клинически дефицит 12 фактора свертывания крови проявляется удлинением времени кровотечения без каких‑либо других симптомов нарушения гемостаза. Выраженность удлинения времени кровотечения зависит от типа наследования генетического дефекта. При доминантном типе дефицит выражен сильнее, при рецессивном — частично компенсируется другими факторами свертывания. Время кровотечения увеличено из‑за замедленной инициации каскадной системы свертывания крови. Склонности к патологическим кровотечениям пациенты не проявляют, поэтому заболевание часто не обнаруживают или обнаруживают случайно при лабораторном обследовании или предоперационном скрининге.
Немного парадоксальным кажется то, что при дефиците фактора свертывания проявляют себя не кровотечения, а тромбозы. Например, Джон Хагеман, по имени которого назван фактор свертывания, впоследствии погиб от тромбоэмболии, которой осложнился перелом костей таза. Современные опубликованные клинические случаи оперативных вмешательств у пациентов с синдромом Хагемана, к счастью, заканчивались благополучно. Тромбофилические нарушения связаны с тем, что XII фактор участвует в разрушении тромбов путем активации калликреин-кининовой системы. Активный калликреин не только запускает генерацию кининов, регулирующих воспаление, сосудистый тонус и болевые реакции, но и превращает неактивный белок плазминоген в активный фермент плазмин (фибринолизин), который и растворяет фибриновую часть тромба. Поэтому с дефицитом фактора Хагемана ассоциированы:
- тромбозы
- мигрирующие тромбофлебиты
- тромбоэмболии
- инфаркты
- спонтанные аборты
Диагноз и прогноз
Кроме лабораторных исследований установить диагноз помогут анамнестические данные: склонность к длительным кровотечениям и тромботическим осложнениям у пациента и его родственников, а также положительный симптом Румпеля — Лееде (появление мелкоточечных кровоизлияний дистально от наложенного на плечо жгута). Прогноз при дефиците фактора Хагемана в большинстве случаев благоприятный, лечение не требуется. Коррекции это состояние требует только в связи с хирургическими вмешательствами. В качестве подготовки к операции может быть назначено переливание небольших доз свежезамороженной плазмы.
Период выведения донорского XII фактора — 48–56 ч. Также при наличии этой коагулопатии следует уделять большее внимание профилактике тромботических осложнений: профилактическая компрессия нижних конечностей, УЗИ-контроль состояния вен нижних конечностей и малого таза, особенно при длительном постельном режиме. В послеоперационном периоде для профилактики тромбозов необходимо назначение низкомолекулярных гепаринов, а для терапии кровотечений — отказ от применения ингибиторов фибринолиза, таких как аминокапроновая и транексамовая кислота.
Редкий клинический случай болезни Хагемана у пациентки кардиологического отделения
Авторы: Ахмедов В.А. 1 , Шадевский В.М. 2 , Судакова А.Н. 2 , Гаус О.В. 1 , Гудалов С.О. 3 , Шустов А.В. 4
1 ФГБОУ ВО ОмГМУ Минздрава России, Омск, Россия
2 ФГБОУ ВО ОмГМУ Минздрава России, Омск
3 БУЗОО «ККД», Омск
4 РГП «Национальный центр биотехнологии», Астана
Болезнь Хагемана — крайне редкое нарушение гемостаза, характеризующееся значительным снижением активности плазменного фактора XII свертывания крови. Представленный клинический случай характеризует выявление данного редкого заболевания — болезни Хагемана у пациентки 58 лет с кардиальной патологией. Пациентка неоднократно поступала в кардиологическое отделение для проведения плановой терапии по поводу ишемической болезни сердца и артериальной гипертензии. Во время одного из поступлений было выявлено удлинение активированного частичного тромбопластинового времени (АЧТВ), что явилось поводом для дальнейшего дообследования. Проведение обследования в условиях Гематологического научного центра позволило выявить наличие редкой патологии коагуляции — болезни Хагемана на основании отсутствия в коагулограмме фактора XII, удлинения АЧТВ и замедления фибринолиза. Особенностью данного случая является то, что заболевание длительное время протекало бессимптомно, усугубляя течение кардиоваскулярной патологии. Особый интерес представляет тот факт, что у пациентки было обнаружено полное отсутствие фактора XII свертывания крови, тогда как в опубликованных ранее наблюдениях описан только дефицит данного фактора.
Ключевые слова: редкий случай, болезнь Хагемана, отсутствие фактора XII, свертывающая система крови, гемостаз, коагулограмма, АЧТВ.
Для цитирования: Ахмедов В.А., Шадевский В.М., Судакова А.Н. и др. Редкий клинический случай болезни Хагемана у пациентки кардиологического отделения. РМЖ. 2018;6(I):46-48.
Rare clinical case of Hageman disease in the patient of the cardiological department
Akhmedov V.A. 1 , Shadevsky V.M. 1 , Sudakova A.N. 1 , Gaus O.V.1, Gudalov S.O. 2 , Shustov A.V. 2
1 Omsk State Medical University
2 “Clinical Cardiology Hospital”, Omsk
Hageman’s disease is an extremely rare disorder of hemostasis, characterized by a significant decrease in the activity of plasma blood coagulation factor XII. The presented case reports the identification of rare Hageman disease in the woman of 58 years old with cardiac pathology. The patient repeatedly admitted to the cardiology department for treatment of ischemic heart disease, arterial hypertension. During one of the visits, an elongation of the activated partial thromboplastin time (APTT) was revealed, which was the cause for further follow-up. Conducting the examination in the conditions of the hematological scientific center revealed a rare pathology of coagulation — Hageman’s disease, based on the absence of factor XII in the coagulogram, prolongation of the APTT and retardation of fibrinolysis. The feature of this case is that the disease has been asymptomatic for a long time, aggravating the course of cardiovascular pathology. Particularly interesting is the fact that the patient was found to have a complete absence of blood coagulation factor XII, whereas in previously published observations only the deficiency of this factor was described.
Key words: rare case, Hageman’s disease, absence of factor XII, coagulating blood system, hemostasis, coagulogram, APTT.
For citation: Akhmedov V.A., Shadevsky V.M., Sudakova A.N. et al. Rare clinical case of Hageman disease in the patient of the cardiological department // RMJ. 2018. № 6(I). P. 46–48.
Представлен клинический случай редкого заболевания — болезни Хагемана — нарушение гемостаза, характеризующееся значительным снижением активности плазменного фактора XII свертывания крови,у пациентки 58 лет с кардиальной патологией.
Болезнь Хагемана (или дефект Хагемана, или дефицит фактора XII свертывания крови) — крайне редкое нарушение гемостаза, характеризующееся значительным снижением активности плазменного фактора XII
(ФXII, фактор Хагемана) свертывания крови [1]. Частота встречаемости болезни Хагемана в популяции не превышает 1 случая на 1 млн населения [2]. В большинстве случаев данный дефект наследуется по рецессивно-аутосомному типу, однако в некоторых семьях выявлено аутосомно-доминантное наследование [1, 2]. По-видимому, синтез фактора XII свертывания крови контролируется двумя аутосомными генами (бимодальный тип наследования). Ген, кодирующий синтез фактора XII свертывания крови, локализуется в 5-й хромосоме [1].
ФXII — сиалогликопротеин (молекулярная масса
около 82 кДа), плазменный профермент, место синтеза которого в организме точно не установлено [3]. ФXII подвержен контактной и ферментной активации, в т. ч. адреналином, калликреином в присутствии отрицательно заряженного высокомолекулярного кининогена, фактором XI, а следовательно, играет ключевую роль в функционировании не только свертывающей, но и калликреин-кининовой, а также фибринолитической систем гемостаза [4].
Система гемостаза филогенетически предназначена для остановки кровотечения при травме и обеспечения жидкого состояния крови при сохранении биологической целостности сосудистой стенки [5]. С современных позиций различают первичный, или сосудисто-тромбоцитарный, и вторичный, или коагуляционный, гемостаз. Первичный гемостаз обусловлен спазмом сосудов и их механической закупоркой агрегатами тромбоцитов, вторичный представляет собой сложный ферментный процесс, в котором участвуют ряд протеолитических ферментов, а также неферментные белковые и фосфолипидные компоненты [1, 5]. Коагуляционный гемостаз может протекать по внешнему и внутреннему механизму, активатором последнего и служит ФXII [2, 4]. Завершающим этапом работы системы гемостаза является фибринолиз — расщепление фибрина под воздействием плазмина, в результате чего происходят разрушение фибринового сгустка и реканализация сосудов после остановки кровотечения [1, 5]. Между процессами свертывания крови и фибринолиза в организме постоянно поддерживается равновесие. Фибринолиз также может протекать по внешнему или внутреннему механизму [2, 4]. Внешний путь активации осуществляется при участии тканевых активаторов, таких как тканевый активатор плазминогена и урокиназа, синтезируемых непосредственно в эндотелии сосудов. Внутренний механизм активации осуществляется благодаря плазменным активаторам (Хагеман-зависимый путь) и активаторам форменных элементов крови (Хагеман-независимый путь). Хагеман-зависимый фибринолиз происходит под влиянием ФXII свертывания крови, который и вызывает превращение неактивного плазминогена в плазмин [1, 2, 5].
Экспериментальные исследования и клинические наблюдения показали, что роль ФXII более значима именно для процессов фибринолиза, нежели для коагуляционного ответа, поэтому при наличии у человека дефекта Хагемана отсутствуют какие-либо геморрагические проявления и отмечается высокая предрасположенность к тромбозам [2, 4, 6].
Впервые дефицит коагуляционного ФXII был выявлен в 1955 г. O.D. Ratnoff и J.E. Colopy, которые представили клиническое наблюдение 37-летнего пациента Джона Хагемана, по имени которого и были названы отсутствующий у него плазменный фактор свертывания и дефект системы гемостаза, обусловленный врожденной недостаточностью этого протеина [7]. Пациент умер от развившейся на фоне длительной иммобилизации тромбоэмболии легочной артерии после перелома тазовых костей.
В настоящее время в мире имеется описание чуть более 100 случаев этого заболевания [8]. Представляем собственное клиническое наблюдение пациентки кардиологического отделения с выявленным дефектом Хагемана.
Пациентка М., 58 лет,
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Дефект Хагемана - диагностика, лечение
Дефект Хагемана врожденное расстройство, передающееся аутосомально-рецессивно, которое происходит благодаря недостатку фактора XII и лишено клинического выражения. Первоначально оно было включено в общее расстройство, носящее название гемофилиоидного синдрома, которое в свою очередь отделилось из общего понятия гемофилии.
В 1954 г. Spaet увидел в нем нозологическую сущность, а в 1955 г. Ratnof обособил это расстройство дав ему имя пациента, на котором он его изучил и описал. Оно называется также недостатком контактного фактора.
До настоящего времени цитируются в литературе 140 случаев. Частота этого дефекта оценивается в 0,01/100 000. Не сообщилось ни одного случая приобретенного дефекта Хагемана.
Патофизиология дефекта Хагемана. Основной дефект этого заболевания состоит в ингибиции генов, индуцирующих синтез Ф. XII и помещающихся на соматических хромозомах. В результате происходит недостаточный синтез Ф. XII, что биологически выражается пертурбацией известных лабораторных тестов, но клинически не дает никаких симптомов.
Этот парадокс, кажущийся необъяснимым, повидимому все же имеет свое объяснение: физиологическое значение Ф. XII гораздо больше при коагуляции in vitro чем in vivo, где его удовлетворительно может замещать Ф. XI. (Последний, для активации in vivo нуждается в коллагене и лишь в минимальном количестве Ф. XII; для активации in vitro, он нуждается в гораздо большем количестве Ф. XII, будучи лишенным коллагена).
Генетическая передача — автосомально рецессивная, причем гены имеют малую пенетрацию, что объясняет отсутствие клинического проявления этого заболевания. Встречается в одинаковой пропорции у мужчин и у женщин и не имеет семейного характера. В двух случаях с геморрагическим синдромом мы отметили единокровность родителей.
Клиника дефекта Хагемана. В данном случае нельзя говорить о болезни в клиническом смысле этого слова, так как 97% носителей этого расстройства не страдают геморрагиями; более того, многие подвергались малым и средним хирургическим вмешательствам, не проявляя склонности к геморрагиям. Следует однако отметить, что существовало несколько случаев, которые, вследствие крупных хирургических вмешательств представляли необычайно сильные геморрагии.
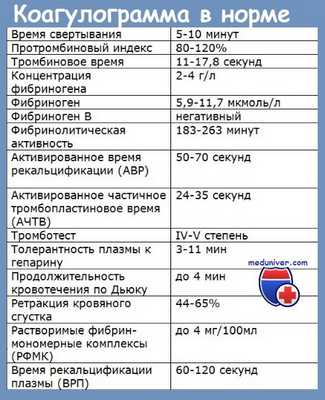
Лабораторное исследование для диагностики дефекта Хагемана показывает:
а) Тестыс аномалийными результатами:
1) ВК удлиненный. Характерным является тот факт, что независимо от того, производятся ли тесты в пробирках из простого стекла или в пробирках из силиконового стекла, получаемые время имеют очень близкие значения (в противоположность недостатку Ф. XI).
2) Т.Н., Р.Т.Т. и РТТК постоянно наммного удлинены. Следует отметить, что Р.Т.Т. и РТТК могут корригироваться тем же способом как и при дефекте Розенталя, что указывает на значительное сходство между Ф. XI и Ф. XII. Тест на толерантность к гепарину показывает сильно пониженную способность коагуляции.
3) Т.С.Р. явно сокращен, указывая на недостаточность расхода протромбина.
4) Т.E.G. показывает значительно удлиненные константы „r" и "k" (преобладает „r").
5) T.G.T. дает точно такой же результат, как и при недостатке Ф. XI. Различие между ними делается лишь тестом Носселя с „celite-plasma".
6) Тесты с нормальными результатами: число тромбоцитов, ВК, капиллярная резистентность, ретракция сгустка, T.Q., Т.Ф. II, Т.Ф. V, Т.Ф. VII+X, Т.Ф. VIII, Т.Ф. IX.
Положительный диагноз дефекта Хагемана ставится только на основании лабораторных тестов. Чаще всего он бывает результатом простой случайности, когда речь идет о больных, исследуемых по совсем иному поводу. Диагноз основывается на результатах вышеуказанных лабораторных тестов. Дифференциальная диагностика производится идентичным образом, как и при недостатке Ф. XI. Индивидуализация в отношении дефекта Розенталя делается при помощи теста Носселя с "celite-plasma".
Эволюция дефекта Хагемана самая благоприятная и лишенная осложнений, из всех известных геморрагических диатезов.
Лечение дефекта Хагемана. Практически нельзя говорить о лечении дефекта Гагемана, так как соответствующие пациенты не страдают в клиническом понимании заболевания.
Для чрезвычайно редких случаев, в которых может появляться то или иное геморрагическое явление (вследствие операции или очень тяжелой травмы), мы применяем такое же лечение, как и при дефекте Розенталя, с той единственной разницей, что имея в виду, что продолжительность полужизни Ф. XII 52 часа, а биологический эффект одной перфузии держится 48 часов, порядок введения должен быть по одной перфузии через 2 дня. Продолжительность лечения зависит от эволюции данного случая, обычно 6—10 дней.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Фибринолитический синдром - диагностика, лечение
Фибринолитический синдром - геморрагический синдром, вызываемый чрезмерной фибринолитической деятельностью, который может появляться в множестве клинических вариантов. В прошлом его включали в категорию плазматических геморрагических диатезов, но в 1959 г. Sherry индивидуализировал его как самостоятельную нозологическую сущность.
Клиника фибринолитического синдрома. С клинической точки зрения, геморрагический синдром может принимать различные аспекты: эпистаксис, крупные экхимозы с контуром географической карты, гастроинтестинальные кровотечения, геморрагии на местах инъекций или пункций, геморрагии после хирургических вмешательств. Вначале эти явления имеют умеренный характер; с течением времени они станивятся все более тяжелыми, так как к ним присоединяются различные недостатки гемостаза, вызываемые самим развитием фибринолитического процесса; в конце концов геморрагический синдром становится таким тяжелым, что ставит в опасность жизнь больного.
Патофизиология фибринолитического синдрома. Нормальное действие механизма фибринолиза обеспечивается динамическим равновесием между активаторами и ингибиторами. Всякий раз когда преобладают активаторы, нарушение равновесия проявляется клинически как фибринолитический синдром; чем больше несоответствие, тем суровее клинический аспект.
Фибринолиз может выступать как самостоятельное расстройство (первичное) или как последствие простой или диссеминированной внутрисосудистой коагуляции (вторичное). Первичный фибринолиз может происходить по поводу роста активаторов плазминогена (спонтанный) или введения вциркуляцию активаторов для лизирования известных тромбов (терапевтически).
Во всех случаях результатом является высвобождение плазмина, который, благодаря своему литическому действию на фибрин, фибриноген, Ф. V, Ф. VIII вызывает геморрагический синдром, описанный в разделе симптоматологии.
Первичный фибринолиз бывает крайне редко (5%); вторичный фибринолиз встречается гораздо чаще.
Лабораторное исследование для диагностики фибринолитического синдрома. Результаты лабораторных тестов представляют большое разнообразие в зависимости от момента когда они производятся и от типа фибринолиза больного (первичного или вторичного). Ниже мы остановимся на тестах первичного фибринолиза, так как вторичный фибринолиз будет представлен в связи с синдромом ДВС.
Т.Н., РТТ и T.Q. могут быть слегка удлиненными (F.D.P. интерферирует с функцией тромбоцитов и с активностью тромбина, а плазма лизирует Ф. V и VIII). Сгустки проб маленькие (мало фибриногена). TLCE значительно сокращен; чем он короче, тем тяжелее синдром. Тест Аструпа (с пластинками фибрина) позволяет выделять каузальный агент фибринолиза: лизокиназа, активатор, плазмин. TEG представляет характерную трассу, вида теннисной ракеты. Тест выявления FDP позитивный (со степенями от + до + + + +).
Дозировка фибриногена дает тем более низкие цифры, чем сильнее фибринолиз. Остальные тесты на гемостаз и коагуляцию дают нормальные результаты.
Положительный клинический диагноз фибринолитического синдрома основывается на следующем: позднее появление кровотечения, картообразный контур экхимозов, кровотечения на месте инъекций и пункций, маленький и хрупкий сгусток, который высвобождает большое количество эритроцитов (когда синдром тяжелый — кровь теряет способность коагулироваться!).
Лабораторные исследования показывают почти нормальные тесты на коагуляцию наряду с позитивными тестами на фибринолиз, что позволяет ставить несомненный диагноз. Дифференциальная диагностика производится по отношению к остальным геморрагическим диатезам. Обстоятельства, при которых возникают кровотечения и лабораторные результаты выясняют неоспоримо диагноз.
Течение и осложнения фибринолитического синдрома. Фибринолитический синдром может иметь очень разнообразную эволюцию. В рамках такой эволюции хронический и острый фибринолитические синдромы находятся на двух крайностей.
Хронический синдром имеет доброкачественную эволюцию и без осложнений. Он может обостряться по поводу хирургического вмешательства, произведенного без антифибринолитической защиты.
Острый или молниеносный синдром имеет драматичную эволюцию. Смерть может наступать до постановки диагноза и назначения лечения. В диагностицированных и леченных по современным методам случаях получаются благоприятные результаты еще в первые 12 часов.
Лечение фибринолитического синдрома относится к острому синдрому и преследует цель прекращения геморрагического синдрома. В качестве эффективных средств можно использовать:
а) Антифибринолитические, которые пресекают механизм фибринолиза; этого можно добиться двумя способами:
1) Антиплазминовое действие: блокирование плазмина, которое осуществляют антиплазмины или протеазовые ингибиторы, двух типов: ингибитор Kunitz, изготовляемый из поджелудочной железы и выпускаемый в продажу под названием Iniprol и ингибитор Frey, изготовляемый из околоушной слюнной железы и выпускаемый в продажу под названием Trasylol (первый в десять раз более активный чем второй).
2) Антиактивирующее действие: блокирование активации плазминогена в плазмин, которое осуществляют синтетические вещества двух типов: с линейной молекулой (ЕАСА) и циклической молекулой (АМСНА) (последнее в 7 раз более активное чем первое).
б) Субституционные: инъецируемый фибриноген и лиофилизированная антигемофилическая плазма, оба содержащие факторы коагуляции, которые в процессе гиперфибринолиза были лизированы в плазме больного и которые мы замещаем при помощи перфузии.
Схема лечения фибринолитического синдрома: мы начинаем с применения Trasylol 1 000 000 Ед в виде медленной перфузии в течение 24 часов. Через час после начала перфузии Trasylol-ом, инъецируется медленно в.в. ЕАСА в дозе 0,3 г/кг веса тела/день, разделенной на 4 приема (по 1 через 6 часов).
Спустя 2 часа от первой инъекции ЕАСА, инъецируется в.в. фибриноген 2 г и продолжается перфузия одного флакона лиофилизированной антигемофилической плазмы. Обычно за 24 часа эффект лечения оказывается благоприятным, так что его следует прервать; если состояние больного требует этого, мы повторяем на следующий день то же лечение. (Внимание! при вторичном фибринолизе все вышеуказанное лечение должно предшествоваться введением гепарина: 40 000 Ед/день, по 10 000 Ед в.в., через 6 часов в течение 2—3 дней).
Антифибринолитическое лечение при ДВС применяется только если во время гепаринового лечения возникает известная степень фибринолиза, требующего такое лечение.
Читайте также:
- Применение соды (бикарбоната натрия) у пациента с отравлением
- Лепра - возбудитель заболевания, лечение и симптомы, диагностика
- Рентгенограмма, КТ при фиброзной дисплазии челюсти
- Морфология туберкулезного орхита. Туберкулез мужских половых органов
- Рентгенограмма, КТ, МРТ при переломе дистального метаэпифиза большеберцовой кости