Дефекты синтеза андрогенов. Причины и диагностика
Добавил пользователь Евгений Кузнецов Обновлено: 30.01.2026
Нарушения биосинтеза андрогенов характеризуются тем, что яички дифференцируются и продуцируют антимюллеров гормон, однако образования андрогенов не происходит. Наружные половые органы сформированы по женскому типу или имеют двойственное строение, но внутренние производные мюллерова протока отсутствуют.
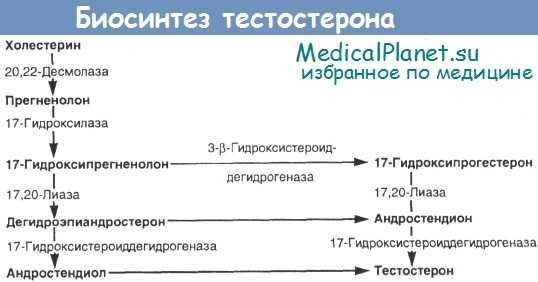
Если снижен уровень тестостерона или его метаболитов, то это заставляет думать о недостаточности ферментов. У детей определить снижение не всегда просто, поскольку и в норме фоновый уровень тестостерона у них невысок, в связи с чем для установления диагноза рекомендуется проведение теста со стимуляцией человеческим хорионическим гонадотропином (ХГ). Схема биосинтеза андрогенов представлена на рисунке.
Дефицит 3-b-гидроксистероиддегидрогеназы (ГСД), 17-b-ГСД, 17-a-гидроксилазы/17,20-лиазы или нарушения в системе ферментов, участвующих в превращении холестерина в прегненолон (врожденная липоидная гиперплазия надпочечников), могут привести к неполной маскулинизации.
Предполагалось, что врожденная липоидная гиперплазия надпочечников обусловлена дефектом в системе ферментов, отщепляющих боковые цепи холестерина на пути его превращения в прегненолон, т.е. 20-а-гидролазы, 20,22-десмолазы или 22-сс-гидроксилазы. Однако недавно были обнаружены мутации в гене, кодирующем белок StAR (англ. steroidogenic acute regulatory protein — белок быстрой регуляции стероидогенеза), который, по-видимому, участвует в транспорте холестерина от наружной к внутренней мембране митохондрий [Tee et al.]. Вероятно, мутации в гене StAR являются главной причиной возникновения этого заболевания.
Половые органы у пациентов данной категории сформированы по женскому типу или имеют двойственное строение. У больных обнаруживаются тяжелые симптомы потери соли. Заболевание наследуется по аутосомно-рецессивному типу, описаны уже более 30 случаев этой патологии, хотя большинство больных погибли в детском возрасте в результате надпочечникового криза.
У одной пациентки, с которой мне недавно пришлось иметь дело, диагноз заболевания был поставлен на основании того, что предыдущий ребенок в этой семье умер в неонатальном периоде и при аутопсии была обнаружена липоидная гиперплазия надпочечников (в них содержались ксантомные клетки, имеющие пенистый вид и заполненные холестерином). Девочку, о которой идет речь, тщательно наблюдали после рождения, и когда появились симптомы потери соли, было назначено эффективное лечение кортикостероидами. К сожалению, на этой стадии заболевания хромосомный анализ не производили и генетический пол девочки определен не был.
Лишь после того как обнаружилось нарушение полового созревания, было установлено, что она имеет кариотип 46XY. Столь неожиданное открытие привело к возникновению у подростка серьезных психологических проблем.
Другим аутосомно-рецессивным расстройством является дефицит 3-b-ГСД, который может привести к гипоспадии или формированию половых органов двойственного строения у лиц генетического мужского пола. Заболевание имеет две формы: с потерей соли и без потери соли. Были клонированы гены изоферментов типа I и типа II, и у ряда пациентов обнаружены мутации в гене 3-b-ГСД типа II [Tajima et al.].
Р450С17 представляет собой особый фермент, опосредующий активность и 17-a-гидроксилазы, и 17,20-лиазы. Обзор соответствующих данных опубликован Yanase и соавт.. Дефицит этого фермента у мужчин обычно проявляется в развитии половых органов двойственного строения, однако в тяжелых случаях недостаточности могут формироваться наружные половые органы женского типа.
Ко времени полового созревания наиболее типичным признаком может быть гинекомастия. Чаще всего у мужчин наблюдается нормальное кровяное давление, хотя оно бывает повышенным вследствие избыточной продукции кортикостерона. Дефект наследуется по аутосомно-рецессивному типу, клонирован соответствующий ген. Сведений о способах пренатальной диагностики в литературе нет.
Дефицит 17-b-ГСД является наиболее частой причиной нарушения биосинтеза тестостерона и распознается по возрастанию уровня андростендиона (или по увеличению соотношения андростендион: тестостерон). Дефект наследуется как аутосомно-рецессивный признак. Фермент катализирует превращение андростендиона в тестостерон в яичках, хотя эта реакция обратима и в экстрагонадных тканях тестостерон может превращаться в андростендион.
Изофермент 17-b-ГСДЗ катализирует процесс в яичках, и кодирующий его ген картирован в области 9q22. У женщин с кариотипом 46XY обнаружены мутации этого гена [Geissler et al.]. Наружные половые органы при рождении обычно женского типа, однако могут иметь слабые или умеренно выраженные признаки двойственности строения. В случае наличия при рождении фенотипически женских половых органов о заболевании можно думать, если пальпируются гонады. Внутренние структуры вольфова протока развиваются нормально, тогда как производные мюллерова протока отсутствуют.
Чаще диагноз удается поставить лишь в период наступления половой зрелости: не появляются менструации, внешний облик пациента приобретает мужские черты, развиваются вторичные мужские половые признаки, хотя у некоторых пациентов имеется гинекомастия. В отдельных случаях половое развитие вначале идет по женскому типу и лишь затем наступает маскулинизация. Пластическая хирургия наружных половых органов может дать хорошие косметические и функциональные результаты, однако бесплодие при данном заболевании неизбежно.
Остается непонятным, почему у этих пациентов в отсутствие тестостерона in Шею развиваются вольфовы протоки. Одной из причин может быть вирилизация вольфовых протоков под влиянием самого андростендиона; кроме того, возможно, что андростендион превращается в тестостерон в плаценте, где высок уровень экспрессии изофермента 17-b-ГСД2. Неясно также, почему вирилизация происходит по достижении пубертатного периода. Согласно одной из гипотез, при высоком уровне циркулирующего андростендиона может происходить его трансформация в тестостерон на периферии под влиянием изоферментов 17-b-ГСД1 или 17-b-ГСД2 или же другого, еще не идентифицированного изофермента.
Каковы бы ни были причины наблюдаемых явлений, если диагноз установлен до наступления половой зрелости, яички можно удалить для предотвращения вирилизации.
Дефекты синтеза андрогенов. Причины и диагностика
Синдром нечувствительности к андрогенам. Причины и диагностика
Существует две формы синдрома: частичная и полная. Какая из них развивается, зависит от остаточной функции андрогеновых рецепторов (АР). Синдром полной нечувствительности к андрогенам характеризуется остановкой развития производных мюллеровых протоков. При этом нет промежуточного типа развития гениталий, хотя налицо признаки тестикулярной феминизации: нормально развитые половые губы, клитор и вход во влагалище. Иногда можно пропальпировать яички в паховых областях.
Если такое случается у ребенка с женским фенотипом, важно уточнить, что же именно пальпируется: 7% новорожденных девочек с паховой грыжей и отсутствием шейки матки при пальпации или вагиноскопии страдают мужским псевдогермафродитизмом, как правило, на фоне синдрома нечувствительности к андрогенам, или страдают смешанной дисгенезией гонад. При синдроме частичной нечувствительности к андрогенам обнаруживают гениталии промежуточного типа с различной степенью развития вольфовых структур и губно-мошоночного сращения.
Патогенез синдрома нечувствительного к андрогенам
Дефект действия андрогенов развивается вследствие мутации гена Андрогеновых рецепторов (АР), нарушающей связывание тестостерона и ДГТ в андроген-чувствительных органах-мишенях. Ген АР подвергается нарушающей его функционирование мутации в локусе Xqll-13. Пострецепторные процессы, осуществляющие эффект андрогенов в тканях-мишенях, не происходят, несмотря на нормальный синтез андрогенов, из-за чего пренатально развивается недостаточная вирилизация наружных гениталий плода.
Описано более 250 мутаций, в том числе полные и частичные делеции, точечные мутации и микроинсерции/микроделеции. Как и предполагалось, набор функциональных дефектов может быть обусловлен любой из них. Некоторые приводят к полной утрате рецепторов клеточных мембран, в то время как другие — к неполноценному синтезу белка вследствие изменений аффинности связывания субстрата. Изменения аффинности могут вносить вклад в ухудшение передачи сигнала, несмотря на наличие нормальных рецепторов клеточных мембран.
При синдроме полной нечувствительности к андрогенам генотип соответствует фенотипу в большинстве случаев. Это, однако, не так при синдроме частичной нечувствительности к андрогенам. Нормальное количество антимюллерова гормона (АМГ) обусловливает редукцию мюллеровых протоков. Однако структуры — производные вольфовых протоков развиваются неправильно вследствие уменьшения или отсутствия связывания тестостерона и ДГТ.
Диагностика синдрома нечувствительного к андрогенам
Концентрация гормонов соответствуют возрастной норме для мужчин. Однако при пробе со стимуляцией ХГЧ повышается содержание как тестостерона, так и ДГТ. Подтверждение диагноза проводят с помощью анализа мутации гена АР, при котором определяют более 95% мутаций, связанных с синдромом полной или частичной нечувствительности к андрогенам, или с помощью определения связывания субстрата с АР клеток кожи мошонки, которое должно быть снижено при этом заболевании.
Для идентификации мюллеровых структур проводят УЗИ. Если эти структуры обнаруживают, диагноз синдрома полной или частичной нечувствительности к андрогенам исключают.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Недостаточность 5а-редуктазы как причина мужского псевдогермафродитизма. Причины и диагностика
При мужском псевдогермафродитизме у пациентов кариотип 46,XY, есть яички, но отмечают неполную маскулинизацию наружных половых органов. Диагностика этого состояния достаточно трудна для клинициста. В отличие от женского псевдогермафродитизма, в большинстве случаев вызываемого врожденной гиперплазией коры надпочечников (ВГКН) и поступлением экзогенных андрогенов, к мужскому псевдогермафродитизму может приводить ряд заболеваний; в большинстве случаев точную причину вообще не удается определить.
Все возможные причины мужского псевдогермафродитизма можно разделить на две большие группы: следствие патологии действия андрогенов или патологии их синтеза. Самые частые причины, связанные с патологией действия андрогенов, — недостаточность 5а-редуктазы, наследуемая по аутосомно-рецессивному типу, и нечувствительность к андрогенам, наследуемая по Х-сцепленному рецессивному типу. Все дефекты синтеза андрогенов наследуются по аутосомно-рецессивному типу. Другая причина — синдром персистирующих мюллеровых протоков.
Он обусловлен дефектом синтеза антимюллерового гормона (АМГ) и тоже наследуется по аутосомно-рецессивному типу.
При недостаточности 5а-редуктазы II типа внешний вид наружных половых органов может быть самым разнообразным: от типично женского до мужского с гипоспадией и/или уменьшением размеров пениса. Характерны клитороподобный фаллос, гипоспадии, раздвоенная мошонка, персистирующий урогенитальный синус, открывающийся в промежность. Часто присутствует вход во влагалище, заканчивающийся слепым карманом. Яички нередко пальпируются в губно-мошоночных складках или паховом канале, хотя иногда их обнаруживают и интраабдоминально.
Многие больные, страдающие недостаточностью 5а-редуктазы, подвергаются вирилизации в раннем подростковом возрасте. Одним из названий болезни, находившихся в обиходе, было «пенис на 12». Раньше таких больных воспитывали как девочек. Во время полового созревания, когда их гениталии подвергались маскулинизации из-за повышения активности фермента I типа, они меняли пол. Понятно, что одним из основных моментов при этом заболевании становится вопрос, по какому полу будет воспитываться ребенок.
Патогенез недостаточности 5а-редуктазы II типа как причина мужского псевдогермафродитизма
Дефект действия андрогенов в данном случае связан с мутацией фермента 5а-редуктазы II типа, отвечающего за превращение тестостерона в более физиологически активный дигидротестостерон (ДГТ) в андроген-чувствительных тканях. Фермент кодируется геном SRD5A2 (другое название — 5а-RD2), расположенным на хромосоме 5. Описано более 40 мутаций на пяти экзонах этого гена. Хотя большинство из них представляет собой замены аминокислот, описаны и другие, например полные делеции, нонсенс-мутации и/или сплайсинг-мутации.
Яички в паховом канале при мужском псевдогермафродитизме
Под влиянием ДГТ происходят маскулинизация наружных гениталий и формированию урогенитального синуса. В результате уменьшения превращения тестостерона в более активный ДГТ развивается неполная маскулинизация. Благодаря нормальному содержанию тестостерона вольфовы структуры развиваются в полноценные семявыносящие протоки. Вследствие нормального синтеза антимюллерового гормона (АМГ) развитие мюллеровых протоков предотвращается.
Диагностика недостаточности 5а-редуктазы II типа как причина мужского псевдогермафродитизма
Диагноз недостаточности 5а-редуктазы часто ставят при рождении. Однако весьма сходный фенотип может развиваться при синдроме нечувствительности к андрогенам. Лабораторные исследования выявляют нормальное или слегка повышенное содержание тестостерона сыворотки крови при сниженном количестве ДГТ. После нагрузки ХГЧ характерно повышение соотношения тестостерон/ДГТ (более 20). Для определения активности 5а-редуктазы II типа используют культивированные фибробласты кожи гениталий, на которых проводят пробу с превращением тестостерона в ДГТ.
При синдроме нечувствительности к андрогенам соотношение тестостерон/ДГТ не повышено.
- Вернуться в оглавление раздела "гинекология"
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Нарушения на любом из этапов ферментативного биосинтеза тестостерона из холестерина могут приводить к изменениям андрогенного профиля. У развивающегося плода мужского пола это может привести к образованию гениталий промежуточного типа. Как и большинство энзимных дефектов, наследуются эти заболевания по аутосомно-рецессивному типу.
Врожденная липоидная гиперплазия надпочечников редкая тяжелая форма врожденной гиперплазии коры надпочечников (ВГКН), заканчивающаяся летально у двух третей пораженных новорожденных детей. Сущность ее заключается в абсолютном дефиците всех стероидных гормонов надпочечников. У новорожденных имеются наружные гениталии женского типа и выраженная потеря соли. При рождении обнаруживают симптомы тяжелой надпочечниковой недостаточности, включающие синдром задержки роста, рвоту, диарею, гипонатриемию и гипокалиемию.
Патогенез и диагностика врожденной липоидной гиперплазии надпочечников. Врожденная липоидная гиперплазия надпочечников может быть обусловлена мутациями двух разных генов. Мутации гена, кодирующего стероидогенныи активный регуляторный протеин, бывают причиной развития этого синдрома в большинстве случаев. Этот белок служит посредником быстрого входа холестерина в митохондрии, являясь экстренным регулятором стероидогенеза.
Реже причиной развития синдрома выступают мутации гена CPY11A, кодирующего фермент расщепления боковой цепи холестерина 20,22-десмолазу. При предварительных лабораторных исследованиях у детей выявляют снижение содержания глюкокортикоидов, минералокортикоидов, тестостерона и ДГТ наряду с уменьшением содержания натрия и калия. Большинство детей погибают до окончания периода новорожденности. Выжившие должны постоянно получать заместительную терапию глюко- и минералокортикоидами.
Недостаточность 3b-гидроксистероиддегидрогеназы как причина нарушения синтеза андрогенов. Одна из форм врожденной гиперплазии коры надпочечников, которая может проявляться как женским, так и мужским псевдогермафродитизмом. Подробно описана в разделе этой главы, посвященном женскому псевдогермафродитизму.
Недостаточность 17a-гидроксилазы как причина нарушения синтеза андрогенов
Недостаточность 17а-гидроксилазы характеризуется снижением количества, вплоть до полного отсутствия, половых гормонов, синтезируемых как гонадами, так и надпочечниками, при одновременном повышении синтеза минералокортикоидных предшественников. У новорожденных мужского пола половые органы развиты по промежуточному типу, в то время как у новорожденных девочек отмечают недоразвитие половых органов.
Заболевание сопровождается артериальной гипертензией различной степени выраженности и гипокалиемией. У девочек недостаточность 17а-гидроксилазы зачастую диагностируют в раннем подростковом возрасте при обследовании по поводу задержки полового развития, отсутствия вторичных половых признаков или первичной аменореи, хотя в некоторых случаях диагностика возможна уже при рождении.

Мутации гена CYP17 могут клинически проявляться в виде недостаточности 17а-гидроксилазы, 17,20-лиазы или их сочетания. При недостаточности 17а-гидроксилазы снижение содержания кортизола стимулирует синтез кортикотропина, и, хотя продукция стероидов повышается, она все равно блокируется на этапе 17а-гидроксилазы. Компенсаторно накапливаются 17-дезоксистероиды, в том числе прегненолон, прогестерон, дезоксикортикостерон и кортикостерон.
Снижение синтеза андрогенов приводит к гипогонадизму. Вследствие минералокортикоидной активности дезоксикортикостерона возникают гипернатриемия и потеря калия, увеличивается объем плазмы крови, развивается артериальная гипертензия. Обычно выявляют гипокалиемию, на фоне которой снижаются количество альдостерона сыворотки крови и активность ренина плазмы.
Диагноз подтверждают при выявлении выраженного повышения содержания 11-дезоксикортикостерона и кортикостерона сыворотки. Количество прегненолона и прогестерона слегка повышено.
Сывороточные концентрации следующих стероидов ничтожны или совсем не определяются: 17а-гидроксипрегненолона, 17а-гидроксипрогестерона, 11-дезоксикортизола, ДГЭА-сульфата, андростендионаитестостерона. Концентрации эстрогенов в сыворотки крови и моче снижены, а кортикотропина, ФСГ и ЛГ — повышены. Выводимые с мочой метаболиты (17а-гидроксикортикостероиды и 17-кетостероиды) имеют недостаточную концентрацию или отсутствуют.
Возможна пренатальная диагностика: определение содержания стероидов надпочечников в амниотической жидкости.
Недостаточность 17,20-десмолазы (лиазы) как причина нарушения синтеза андрогенов
При этом синдроме блокируются пути превращения 21-углеродных стероидов (17-гидроксипрегненолон и 17-гидроксипрогестерон) в 19-углеродные стероиды, ДГЭА-С и андростендион соответственно. Отмечают также снижение количества андрогенов, тестостерона и эстрадиола. Блокада может быть как полной, так и частичной, в зависимости от этого могут различаться клинические симптомы.
Как уже было сказано выше, ген CYP17 кодирует 17а-гидроксилазно-17,20-лиазный комплекс, а его дефекты могут приводить к недостаточности 17,20-десмолазы (лиазы). Лабораторно выявляют повышение концентрации 17-гидроксипрегненолона сыворотки, 17-гидроксипрогестерона, нормальное содержание ФСГ при повышении ЛГ, снижение тестостерона и эстрадиола, ДГЭА-сульфата, андростендиона, а также нормальное или слегка повышенное содержание прегненолона и прогестерона.
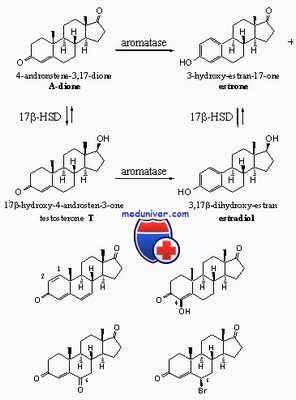
Недостаточность 17b-гидроксистероиддегидрогеназы как причина нарушения синтеза андрогенов
Диагноз этого заболевания зачастую ставят в период полового созревания лицам с мужским генотипом. Они могут воспитываться как девочки и обращаться с жалобами на отсутствие менструаций и гирсутизм, или как мальчики, и тогда обращаться с жалобами на гинекомастию и недоразвитие половых органов. У пораженных лиц мужского пола показатели вирилизации, включая клиторомегалию вплоть до микрофалоса и развитие вторичных мужских половых признаков в период полового созревания, очень напоминают таковые при недостаточности 5а-редуктазы. Все больные бесплодны.
Фермент 17b-гидроксистероиддегидрогеназа, или 17-кетостероидредуктаза, катализирует превращение андростендиона в тестостерон в яичках. Мутации гена изофермента 17b-гидроксистероиддегидрогеназы типа 3 (HSD17B3), расположенного на хромосоме 9q22, приводят к недостаточному синтезу тестостерона. Большинство их представлено миссенс/нонсенс-мутациями. Существенной корреляции между генотипом и фенотипом не выявляют.
Развитие гениталий по промежуточному типу обычно выявляют при рождении. Чаще всего имеются кли-торомегалия, сращение губно-мошоночных складок и слепой влагалищный карман. Яички нередко пальпируются в паховых каналах или губно-мошоночных складках, хотя иногда могут быть расположены и в брюшной полости. Как и при других формах мужского псевдогермафродитизма, внутренние отделы мочеполового тракта развиты нормально.
Есть придатки яичка, семявыносящие протоки, семенные пузырьки, семявыбрасывающие протоки. Предстательная железа и производные мюллеровых протоков отсутствуют.
Характерной лабораторной находкой бывает повышенное соотношение андростендион/тестостерон, образующееся в результате увеличения количества андростендиона и снижения тестостерона. Возможна пренатальная диагностика у потомства пораженных пациентов, если у последних выявлена специфическая мутация.
Адреногенитальный синдром ( ВДКН , Врожденная дисфункция коры надпочечников )
Адреногенитальный синдром — наследственное заболевание надпочечников, при котором вследствие функциональной несостоятельности ферментов нарушается стероидогенез. Проявляется вирилизацией гениталий, маскулиноподобным телосложением, недоразвитием груди, гирсутизмом, акне, аменореей или олигоменореей, бесплодием. В ходе диагностики определяют уровни 17-гидроксипрогестерона, 17-кетостероидов, андростендиона, АКТГ, проводят УЗИ яичников. Пациенткам назначают заместительную гормонотерапию глюкокортикоидами и минералокортикоидами, эстрогены в комбинации с андрогенами или прогестинами нового поколения. При необходимости выполняют пластику половых органов.
МКБ-10
Общие сведения
Адреногенитальный синдром, или врожденная дисфункция (гиперплазия) коры надпочечников, — наиболее частое из наследуемых заболеваний. Распространенность патологии отличается у представителей разных национальностей. Классические варианты АГС у лиц европеоидной расы встречаются с частотой 1:14 000 младенцев, в то время как у эскимосов Аляски этот показатель составляет 1:282.
Существенно выше заболеваемость у евреев. Так, неклассическую форму адреногенитального расстройства выявляют у 19% лиц еврейской национальности группы ашкенази. Патология передается по аутосомно-рецессивному типу. Вероятность рождения ребенка с таким синдромом при носительстве патологического гена у обоих родителей достигает 25%, в браке носителя и больного — 75%. Если один из родителей имеет полноценные ДНК, клинические проявления синдрома у детей не развиваются. При наличии АДС у отца и матери ребенок также будет болен.
Причины
У больных с наследуемой гиперплазией надпочечников генетический дефект проявляется несостоятельностью ферментных систем, участвующих в секреции стероидных гормонов. В 90-95% случаев патология возникает при повреждении гена, который отвечает за синтез 21-гидроксилазы — фермента, влияющего на образование кортизола. В остальных клинических случаях вследствие дефекта ДНК нарушается производство других ферментов, обеспечивающих стероидогенез, — StAR/20,22-десмолазы, 3-β-гидрокси-стероиддегидрогеназы, 17-α-гидроксилазы/17,20-лиазы, 11-β-гидроксилазы, P450-оксидоредуктазы и синтетазы альдостерона.
У пациентов с признаками вирилизирующего синдрома вместо активного гена CYP21-B в коротком плече 6-й аутосомы расположен функционально несостоятельный псевдоген CYP21-A. Структура этих участков ДНК-цепи во многом гомологична, что повышает вероятность конверсии генов в мейозе с перемещением участка нормального гена на псевдоген или делецию CYP21-B.
По-видимому, именно этими механизмами объясняется существование скрытых форм болезни, дебютирующих в пубертате или постпубертатном периоде. В таких случаях клинические признаки патологии становятся заметными после нагрузок, истощающих кору надпочечников: тяжелых болезней, травм, отравлений, радиационных воздействий, длительного периода интенсивной работы, психологически напряженных ситуаций и т. д.
Патогенез
В основе механизма развития наиболее распространенного варианта адреногенитального синдрома с дефектом CYP21-B-гена лежит принцип обратной связи. Ее начальным звеном становится дефицит стероидов — кортизола и альдостерона. Несостоятельность процессов гидроксилирования сопровождается неполным переходом 17-гидроксипрогестерона и прогестерона в 11-дезоксикортизол и дезоксикортикостерон. В результате снижается секреция кортизола, а для компенсации этого процесса в гипофизе усиливается синтез АКТГ — гормона, вызывающего компенсаторную гиперплазию коры надпочечников для стимуляции выработки кортикостероидов.
Параллельно возрастает синтез андрогенов и появляются видимые признаки их влияния на чувствительные ткани и органы. При умеренном снижении активности фермента минералокортикоидная недостаточность не развивается, поскольку потребность организма в альдостероне почти в 200 раз ниже по сравнению с кортизолом. Только глубокий дефект гена вызывает тяжелую клиническую симптоматику, которая проявляется с раннего возраста. Патогенез развития заболевания при нарушении структуры других участков ДНК аналогичен, однако пусковым моментом являются нарушения в других звеньях стероидогенеза.
Классификация
Систематизация различных форм вирилизирующей гиперплазии надпочечников основана на особенностях клинической картины заболевания, выраженности генетического дефекта и времени проявления первых патологических признаков. Тяжесть расстройства напрямую связана со степенью повреждения ДНК. Специалисты в сфере эндокринологии различают следующие виды адреногенитального синдрома:
- Сольтеряющий. Самый тяжелый вариант патологии, проявляющийся в первый год жизни ребенка грубыми нарушениями строения наружных половых органов у девочек и их увеличением у мальчиков. Активность 21-гидроксилазы составляет не более 1%. Значительное нарушение стероидогенеза приводит к выраженным соматическим нарушениям — рвоте, поносу, судорогам, чрезмерной пигментации кожи. Без лечения такие дети умирают в раннем возрасте.
- Простой вирильный. Течение заболевания менее тяжелое, чем при сольтеряющем варианте. Преобладают проявления неправильного развития гениталий у младенцев женского пола, увеличение их размеров у мальчиков. Признаки надпочечниковой недостаточности отсутствуют. Уровень активности 21-гидроксилазы снижен до 1-5%. С возрастом у пациентов нарастают признаки вирилизации вследствие стимулирующего действия андрогенов.
- Неклассический (постпубертатный). Наиболее благоприятная форма АГС, явные признаки которой возникают в период полового созревания и в репродуктивном возрасте. Наружные половые органы имеют нормальное строение, может быть увеличен клитор у женщин и половой член у мужчин. Функциональность 21-гидроксилазы снижена до 20-30%. Заболевание выявляется случайно при обследовании в связи с бесплодием или нарушениями менструальной функции.
Сольтеряющий и простой вирильный виды адреногенитальных расстройств относят к категории антенатальной патологии, формирующейся внутриутробно и проявляющейся с момента рождения. При дефекте строения других генов наблюдаются более редкие варианты заболевания: гипертензивные — классический (врожденный) и неклассический (поздний), гипертермический, липидный, с ведущими проявлениями гирсутизма.
Симптомы
Сольтеряющий и простой вирильный
При антенатальных формах заболевания основным клиническим симптомом является видимая вирилизация гениталий. У новорожденных девочек обнаруживаются признаки женского псевдогермафродитизма. Клитор большой по размерам или имеет пенисообразную форму, преддверие влагалища углублено, сформирован урогенитальный синус, большие и малые половые губы увеличены, промежность высокая. Внутренние половые органы развиты нормально.
У младенцев-мальчиков увеличен половой член и гиперпигментирована мошонка. Кроме того, при сольтеряющем адреногенитальном расстройстве выражена симптоматика надпочечниковой недостаточности с тяжелыми, зачастую несовместимыми с жизнью соматическими нарушениями (понос, рвота, судороги, обезвоживание и др.), которые проявляются с 2-3-недельного возраста. У девочек с простым вирильным АГС по мере взросления признаки вирилизации усиливаются, формируется диспластическое телосложение.
Из-за ускорения процессов окостенения пациентки отличаются невысоким ростом, широкими плечами, узким тазом, короткими конечностями. Трубчатые кости массивные. Половое созревание начинается рано (до 7 лет) и протекает с развитием вторичных мужских половых признаков. Отмечается увеличение клитора, снижение тембра голоса, нарастание мышечной силы, формирование типичной для мужчин формы перстневидного хряща щитовидной железы. Грудь не растет, менархе отсутствует.
Неклассический
Менее специфичны клинические симптомы при неклассических формах вирилизирующего синдрома, возникшие в пубертате и после стрессовых нагрузок (выкидыша на ранних сроках беременности, медицинского аборта, операции и др.). Обычно пациентки вспоминают, что у них еще в младшем школьном возрасте появилось небольшое оволосение в подмышечных впадинах и на лобке. В последующем развились признаки гирсутизма с ростом стержневых волос над верхней губой, по белой линии живота, в области грудины, в сосково-ареолярной зоне.
Женщины с АГС предъявляют жалобы на стойкую угревую сыпь, пористость и повышенную жирность кожи. Менархе наступает поздно — к 15-16 годам. Менструальный цикл неустойчив, интервалы между менструациями достигают 35-45 дней и более. Кровянистые выделения во время месячных скудные. Молочные железы небольшие. Клитор несколько увеличен. Такие девушки и женщины могут иметь высокий рост, узкий таз, широкие плечи.
По наблюдениям специалистов в сфере акушерства и гинекологии, чем позже развиваются адреногенитальные расстройства, тем менее заметны внешние признаки, характерные для мужчин, и тем чаще ведущим симптомом становится нарушение месячного цикла. При более редких генетических дефектах пациентки могут жаловаться на повышение артериального давления или, наоборот, гипотонию с низкой работоспособностью и частыми головными болями, гиперпигментацию кожи с минимальными симптомами вирилизации.
Осложнения
Основным осложнением адреногенитального синдрома, по поводу которого пациентки обращаются к акушерам-гинекологам, является стойкое бесплодие. Чем раньше проявилось заболевание, тем меньше вероятность забеременеть. При значительной ферментной недостаточности и клинических проявлениях простого вирилизирующего синдрома беременность вообще не наступает.
У забеременевших пациенток с пубертатными и постпубертатными формами заболевания возникают самопроизвольные выкидыши на раннем сроке. В родах возможна функциональная истмико-цервикальная недостаточность. Такие женщины более склонны к возникновению психоэмоциональных расстройств — склонности к депрессии, суицидальному поведению, проявлениям агрессии.
Диагностика
Постановка диагноза при антенатальных типах АГС с характерными изменениями половых органов не представляет сложности и проводится сразу после родов. В сомнительных случаях применяют кариотипирование для подтверждения женского кариотипа (46ХХ), молекулярно-генетические тесты. Большее значение диагностический поиск приобретает при позднем клиническом дебюте или скрытом течении с минимальными внешними проявлениями вирилизации. В подобных ситуациях для выявления адреногенитального синдрома используют следующие лабораторные и инструментальные методы:
- Уровень 17-ОН-прогестерона. Высокая концентрация 17-гидроксипрогестерона, который является предшественником кортизола — ключевой признак недостаточности 21-гидроксилазы. Его содержание увеличено в 3-9 раз (от 15 нмоль/л и выше).
- Стероидный профиль (17-КС). Повышение уровня 17-кетостероидов в моче у женщин в 6-8 раз свидетельствует о высоком содержании андрогенов, производимых корой надпочечников. При выполнении преднизолоновой пробы концентрация 17-КС уменьшается на 50-75%.
- Содержание андростендиона в сыворотке крови. Повышенные показатели этого высокоспецифичного метода лабораторной диагностики подтверждают усиленную секрецию предшественников мужских половых гормонов.
- Уровень АКТГ в крови. Для классических форм заболевания характерна компенсаторная гиперсекреция адренокортикотропного гормона передней долей гипофиза. Поэтому при синдроме вирилизирующей дисфункции показатель повышен.
- УЗИ яичников. В корковом веществе определяются фолликулы на разных стадиях созревания, не достигающие преовуляторных размеров. Яичники могут быть несколько увеличены, однако разрастания стромы не наблюдается.
- Измерение базальной температуры. Температурная кривая типична для ановуляторного цикла: первая фаза растянута, вторая укорочена, что обусловлено недостаточностью желтого тела, которое не образуется из-за отсутствия овуляции.
Для сольтеряющего варианта АГС также характерна повышенная концентрация ренина в плазме крови. Дифференциальная диагностика адреногенитальных расстройств, возникших в пубертатном и детородном возрасте, проводится с синдромом поликистозных яичников, овариальными андробластомами, андростеромами надпочечников, вирильным синдромом гипоталамического происхождения и конституциональным гирсутизмом. В сложных случаях к диагностике привлекают эндокринологов, урологов, врачей-генетиков.
Лечение адреногенитального синдрома
Основным способом коррекции вирильной дисфункции надпочечников является заместительная гормональная терапия, восполняющая дефицит глюкокортикоидов. Если у женщины со скрытым АГС нет репродуктивных планов, кожные проявления гиперандрогении незначительны и месячные ритмичны, гормоны не применяют. В остальных случаях выбор схемы лечения зависит от формы эндокринной патологии, ведущей симптоматики и степени ее выраженности. Зачастую назначение глюкокортикоидных препаратов дополняют другими медикаментозными и хирургическими методами, подобранными в соответствии с конкретной терапевтической целью:
- Лечение бесплодия. При наличии планов по деторождению женщина под контролем андрогенов крови принимает глюкокортикоиды до полного восстановления овуляторного месячного цикла и наступления беременности. В резистентных случаях дополнительно назначают стимуляторы овуляции. Во избежание выкидыша гормонотерапию продолжают до 13-й недели гестационного срока. В I триместре также рекомендованы эстрогены, во II-III — аналоги прогестерона, не обладающие андрогенным эффектом.
- Коррекция нерегулярных месячных и вирилизации. Если пациентка не планирует беременность, но жалуется на расстройство менструального цикла, гирсутизм, угри, предпочтительны средства с эстрогенным и антиандрогенным эффектом, оральные контрацептивы, содержащие гестагены последнего поколения. Терапевтический эффект достигается за 3-6 месяцев, однако по окончании лечения при отсутствии заместительной гормонотерапии признаки гиперандрогении восстанавливаются.
- Лечение врожденных форм АГС. Девочкам с признаками ложного гермафродитизма проводят адекватную гормонотерапию и выполняют хирургическую коррекцию формы половых органов — клитеротомию, интроитопластику (вскрытие урогенитального синуса). При сольтеряющих адреногенитальных расстройствах кроме глюкокортикоидов под контролем рениновой активности назначают минералокортикоиды с увеличением терапевтических доз при возникновении интеркуррентных заболеваний.
Определенные сложности в ведении пациентки возникают в тех случаях, когда заболевание не диагностировано в акушерском стационаре, и девочка с выраженной вирилизацией гениталий регистрируется и воспитывается как мальчик. При решении о восстановлении женской половой идентичности хирургическую пластику и гормонотерапию дополняют психотерапевтической поддержкой. Решение о сохранении гражданского мужского пола и удалении матки с придатками принимается в исключительных случаях по настоянию больных, однако такой подход считается ошибочным.
Прогноз и профилактика
Прогноз при своевременном обнаружении адреногенитального синдрома и адекватно подобранной терапии благоприятный. Даже у пациенток со значительной вирилизацией гениталий после пластической операции возможна нормальная половая жизнь и естественные роды. Заместительная гормонотерапия при любой форме АГС способствует быстрой феминизации — развитию грудных желез, появлению месячных, нормализации овариального цикла, восстановлению генеративной функции. Профилактика заболевания осуществляется на этапе планирования беременности.
Если в роду наблюдались случаи подобной патологии, показана консультация генетика. Проведение пробы с АКТГ обоим супругам позволяет диагностировать гетерозиготное носительство или скрытые формы адреногенитального расстройства. При беременности синдром может быть обнаружен по результатам генетического анализа клеток хорионических ворсин или содержимого околоплодных вод, полученных методом амниоцентеза. Неонатальный скрининг, проводимый на 5-е сутки после родов, направлен на выявление повышенной концентрации 17-гидропрогестерона для быстрого выбора терапевтической тактики.
Читайте также:
- Показания для операции при несостоятельности тотального эндопротеза голеностопного сустава (операции спасения)
- Период новорожденности при многоплодии (многоплодной беременности).
- Кардиальный вариант гипотонического синдрома. Сердечные проявления гипотонии
- Операции при осложненных дуоденальных язвах. Особенности
- Кости при синдроме Кушинга - лучевая диагностика