Дефицит кислой мальтазы. Болезнь Мак Ардла - дефицит миофосфорилазы.
Добавил пользователь Morpheus Обновлено: 29.01.2026
Болезнь Маркиафавы-Микели - достаточно редкая форма приобретенной гемолитической анемии, характеризующаяся нарушением структуры эритроцитов, нейтрофилов и тромбоцитов, сопровождающаяся признаками внутрисосудистого разрушения эритроцитов. При этом очень часто наблюдаются увеличение гемоглобина, гемосидеринурия (гемосидерин в моче), повышение свободного гемоглобина плазмы. Болезнь нередко осложняется тромбозами периферических вен и сосудов внутренних органов.
Болезнь была подробно описана в 1928 г. Markiafava под названием «гемолитическая анемия с постоянной гемосидеринурией», затем в том же году Micheli и названа болезнью Маркиафавы - Микели.
Общее распространенное название заболевания «пароксизмальная ночная гемоглобинурия» (ПНГ).
Данное название мало соответствует сути заболевания, так как при этой болезни нет ни настоящих пароксизмов, ни обязательной гемоглобинурии.
Внутрисосудистое разрушение эритроцитов с гемосидеринурией, кроме болезни Маркиафавы – Микели, наблюдается при ряде других заболеваний. Оно обнаруживается при многих формах аутоиммунной гемолитической анемии как с тепловыми, так и с Холодовыми антителами, особенно при формах с тепловыми гемолизинами; постоянное внутрисосудистое разрушение эритроцитов выявляется при некоторых формах наследственных гемолитических анемий, связанных с нарушением структуры мембраны эритроцита.
Болезнь Маркиафавы - Микели относится к числу редких форм гемолитической анемии.
Что провоцирует / Причины Болезни Маркиафавы-Микели:
В настоящее время не вызывает сомнений, что при болезни Маркиафавы - Микели причиной повышенного разрушения эритроцитов является их дефект. Это было доказано при переливаниях донорских эритроцитов больным и эритроцитов больных здоровым людям.
Патогенез (что происходит?) во время Болезни Маркиафавы-Микели:
При болезни Маркиафавы - Микели поражаются не только эритроциты, но и лейкоциты и тромбоциты. Уменьшение количества этих форменных элементов связано, с одной стороны, с некоторым уменьшением их производства, с другой - с нарушением их структуры и ускоренным разрушением. Было доказано, что тромбоциты и лейкоциты больных синдромом Маркиафавы - Микели обладают повышенной чувствительностью к воздействию комплемента. Они во много раз чувствительнее к действию изоагглютининов, чем донорские тромбоциты и лейкоциты. Другими словами, тромбоциты и лейкоциты имеют тот же дефект мембраны, что и эритроциты.
На поверхности эритроцитов, лейкоцитов и тромбоцитов не удается обнаружить иммуноглобулинов самыми чувствительными методами и таким образом показать принадлежность болезни Маркиафавы - Микели к группе аутоагрессивных заболеваний.
Убедительно доказано существование двух самостоятельных популяций эритроцитов при болезни Маркиафавы Микели. Наиболее стойкие клетки у здорового человека, ретикулоциты, оказываются наиболее хрупкими при болезни Маркиафавы - Микели.
Идентичность поражения мембраны эритроцитов, нейтрофилов и тромбоцитов указывает на то, что патологическую информацию, скорее всего, получает клетка, следующая за стволовой: общая клетка – предшественница миелопоэза.
Описаны единичные случаи развития быстрого лейкоза на фоне этой болезни.
Главную роль в механизме развития тромботических осложнений приписывают внутрисосудистому распаду эритроцитов и стимуляции свертывания факторами, освобождающимися из клеток во время их распада. Было доказано, что ретикулоциты содержат большое количество факторов, способствующих свертыванию крови.
При болезни Маркиафавы - Микели разрушаются в основном ретикулоциты; может быть, этим следует объяснить высокую частоту тромботических осложнений при болезни Маркиафавы - Микели и сравнительную редкость таких осложнений при других формах гемолитических анемий с выраженным внутрисосудистым гемолизом.
Симптомы Болезни Маркиафавы-Микели:
Болезнь чаще начинается внезапно. Больной жалуется на слабость, недомогание, головокружение. Иногда больные обращают внимание на небольшую желтизну склер. Обычно наиболее распространенной первой жалобой является жалоба на боли: сильные головные боли, боли в области живота. Возможно бессимптомное течение заболевания, и тогда лишь склонность к повышенному тромбообразованию заставляет больного обратиться к врачу. Гемоглобинурия редко бывает первым симптомом болезни.
Приступы боли в животе - один из характерных признаков болезни Маркиафавы -Микели. Локализация боли может быть самой различной. Описаны операции в связи с подозрением на острый аппендицит, язву желудка, желчнокаменную болезнь, вплоть до удаления части желудка у таких больных. Вне криза болей в животе, как правило, не бывает. Нередко к болям в животе присоединяется рвота. Вероятно, боли в животе связаны с тромбозами мелких мезентериальных сосудов. Нередко наблюдаются тромбозы периферических сосудов. Тромбофлебит встречается у 12% больных с болезнью Маркиафавы - Микели, как правило, при этом поражаются вены конечностей. Описаны тромбозы сосудов почек. Тромботические осложнения - наиболее частая причина смерти при болезни Маркиафавы – Микели.
При обследовании больного выявляются бледность с небольшим желтушным оттенком, одутловатость лица, иногда излишняя полнота. Небольшое увеличение селезенки возможно, но не обязательно. Печень нередко увеличена, хотя это также не является специфическим признаком болезни.
Болезнь Маркиафавы - Микели сопровождается признаками внутрисосудистого гемолиза, в первую очередь повышением свободного гемоглобина плазмы, наблюдаемым почти у всех больных. Однако выраженность такого повышения различна и зависит от того, в какой период болезни проводилось исследование. В период криза этот показатель значительно возрастает, увеличивается также количество метальбумина плазмы. Уровень свободного гемоглобина зависит и от содержания гаптотлобина, фильтрации гемоглобина в почках, скорости разрушения комплекса гемоглобин - гаптоглобин.
При прохождении через канальцы почек гемоглобин частично разрушается, откладывается в эпителии канальцев, что приводит к выделению с мочой гемосидерина у большинства больных. Это важный признак болезни. Иногда гемосидеринурия выявляется не сразу, лишь в процессе динамического наблюдения за больным. Следует также отметить, что гемосидерин выделяется с мочой при ряде других заболеваний.
Диагностика Болезни Маркиафавы-Микели:
Содержание гемоглобина у больных с болезнью Маркиафавы - Микели колеблется от 30 до 50 г/л в период обострения, до нормы - в период улучшения. Содержание эритроцитов снижается соответственно снижению гемоглобина. Цветовой показатель долго остается близким к единице. Если больной теряет много железа с мочой в виде гемосидерина и гемоглобина, то содержание железа постепенно падает. Низкий цветовой показатель наблюдается приблизительно у половины больных. Иногда повышен уровень гемоглобина F, особенно при обострении.
У значительной части больных содержание ретикулоцитов бывает повышенным, но сравнительно невысоким (2-4%). Иногда в эритроцитах обнаруживаются точечные дефекты. Количество лейкоцитов при болезни Маркиафавы – Микели в большинстве случаев снижено. У многих больных количество лейкоцитов составляет 1,5-3,0 Ч 109/л, но иногда снижается до очень низких цифр (0,7-0,8 Ч 109/л). Лейкопения у большинства больных обусловлена уменьшением количества нейтрофилов. Однако иногда при болезни Маркиафавы - Микели содержание лейкоцитов бывает нормальным и редко повышенным до 10-11 Ч 109 г/л.
При болезни Маркиафавы - Микели снижается фагоцитарная активность нейтрофилов. Тромбоцитопения тоже встречается часто, однако снижения агрегации не наблюдается. Вероятно, с этим связана редкость геморрагических осложнений, хотя количество тромбоцитов иногда падает до очень низких цифр (10-20 Ч 109/л). Обычно у большинства больных содержание тромбоцитов колеблется от 50 до 100 Ч 109/л. Нормальное количество тромбоцитов не исключает болезни Маркиафавы - Микели.
Исследование костного мозга выявляет в основном признаки гемолитической анемии -раздражение красного ростка при нормальном количестве миелокариоцитов. У ряда больных несколько снижено количество мегакариоцитов.
Уровень железа сыворотки при болезни Маркиафавы - Микели зависит от стадии болезни, выраженности внутрисосудистого разрушения эритроцитов и активности кроветворения. В ряде случаев болезнь Маркиафавы - Микели начинается с признаков гипоплазии. Запасы железа в организме у больного зависят, с одной стороны, от потери
железа с мочой, с другой - от интенсивности кроветворения. В частности, это не позволяет считать дефицит железа диагностическим признаком болезни Маркиафавы -Микели.
Болезнь Маркиафавы - Микели может протекать по-разному. П–и этом самочувствие больных вне криза не страдает, содержание гемоглобина около 8090 г/л.
При другом, также типичном, варианте общее состояние больных вне криза нарушается значительно больше. Уровень гемоглобина постоянно низкий - 40-50 г/л. За все время болезни может не быть черной мочи, а если и бывает, то после трансфузии плазмы или свежих неотмытых эритроцитов. Кроме этих двух вариантов, существует ряд переходных форм, когда вначале имеются гемолитические кризы, а затем при прогрессировании анемии они сглаживаются. У некоторых больных тяжелые гемолитические кризы следуют один за другим, приводят к выраженной постоянной анемии. Кризы часто сопровождаются тромботическими осложнениями. У некоторых больных картина болезни в основном определяется тромбозами, а уровень гемоглобина держится около 9,5-100 г/л.
У части больных болезнь Маркиафавы - Микели начинается с апластической анемии.
Дифференциальная диагностика проводится с рядом заболеваний в зависимости от того, на какой симптом болезни врач обратил внимание.
Если у больного бывает черная моча, а в лаборатории не вызывает затруднений идентификация гемосидерина в моче, то диагностика облегчается. Круг болезней с внутрисосудистым разрушением эритроцитов ограничен, поэтому правильный диагноз ставят довольно быстро.
Чаще, однако, внимание врача привлекают другие симптомы болезни: боли в животе, тромбозы периферических сосудов, анемия. Нередко это заставляет заподозрить злокачественное новообразование желудочно-кишечного тракта. У таких больных проводят рентгенологические исследования желудка, кишечника, а если видят красную мочу, то исследуют почки.
Нередко врачи обращают внимание на высокий белок в моче и предполагают какое-либо заболевание почек. При этом не учитывается, что выраженная протеинурия, темный цвет мочи и отсутствие в ней эритроцитов чаще бывают при гемоглобинуриях, поскольку гемоглобин - это тоже белок.
В этих случаях целесообразно выполнить бензидиновую пробу Грегерсена с мочой, если в ней нет эритроцитов. Кроме гемоглобина, положительную пробу Грегерсена может обусловить миоглобин, однако миоглобинурии бывают значительно реже, чем гемоглобинурия, и при них содержание гемоглобина не снижается.
Выраженная панцитопения при болезни Маркиафавы - Микели приводит к диагностике апластической анемии. Однако для этой группы заболеваний не характерны повышение количества ретикулоцитов, раздражение красного ростка костного мозга, внутрисосудистое разрушение эритроцитов.
Гораздо труднее отличить от болезни Маркиафавы - Микели с гемолизиновой формой аутоиммунную гемолитическую анемию. Они практически почти не различаются по
клинической картине, однако количество лейкоцитов при болезни Маркиафавы - Микели чаще снижено, а преднизолон практически неэффективен, тогда как при аутоиммунной гемолитической анемии лейкоциты чаще повышены, а при ее гемолизиновых формах преднизолон часто дает хороший эффект.
Диагностике помогают выявление гемолизинов сыворотки по стандартной методике и по модификациям сахарозной пробы, а также выявление на поверхности эритроцитов антиэритроцитарных антител.
Выделение с мочой гемосидерина в сочетании с болями в животе, гипохромной анемией, тромбоцитопенией наблюдается иногда при тяжелой свинцовой интоксикации. Однако в этих случаях бывает полиневритический синдром, отсутствующий при болезни Маркиафавы - Микели. Кроме того, для болезни Маркиафавы – Микели характерны положительные сахарозная проба и проба Хема. При свинцовом отравлении они отрицательные. Свинцовое отравление сопровождается резким повышением содержания 6-аминолевулиновой кислоты и копропорфирина в моче.
Лечение Болезни Маркиафавы-Микели:
Методов лечения, направленных на механизм развития заболевания, не существует. Глубина анемии при болезни Маркиафавы - Микели определяется выраженностью, с одной стороны, гипоплазии, а с другой - разрушением эритроцитов. Тяжелое общее состояние больного и низкий уровень гемоглобина служат показателями для переливания крови.
Число переливаний крови определяется состоянием больного, быстротой повышения уровня гемоглобина. Многие больные постоянно нуждаются в переливаниях крови с интервалами от 3–4 дней до нескольких месяцев. Вначале больные хорошо переносят процедуры, но после месяцев или лет болезни у них нередко развиваются тяжелые реакции даже на тщательно отмытые эритроциты. Это требует подбора эритроцитов по непрямой пробе Кумбса.
Часть больных получали неробол по 5 мг 4 раза в день с некоторым эффектом. Неробол следует применять в течение нескольких месяцев под контролем функции печени в связи с возможностью развития холестатического гепатита. Действие препарата пролонгированного действия (ретаболила) слабее.
Оксиметалон значительно менее токсичен, чем неробол, назначается по 150-200 мг/сут.; холестатический эффект больших доз препарата значительно меньше, чем у неробола. Анаполон применяется по 150-200 мг/сут. в течение 3-4 месяцев.
В связи с повышенной способностью к образованию перекисей непредельных жирных кислот в мембране эритроцитов больных болезнью Маркиафавы - Микели возникает вопрос о применении препаратов токоферола. Витамин Е обладает антиоксидантными свойствами, способен противостоять действию окислителей. В дозе 3-4 мл/сут. (0,15-0,2 мкг ацетата токоферола) препарат оказывает определенное антиоксидантное действие. В частности, его можно использовать для профилактики гемолитического криза при применении препаратов железа.
Препараты железа показаны при его значительной потере и выраженном дефиците.
Для борьбы с тромбозами при болезни Маркиафавы - Микели используют гепарин, чаще в небольших дозах (5000 ЕД 2-3 раза в день в кожу живота), а также антикоагулянты непрямого действия.
Прогноз
Продолжительность жизни больных колеблется от 1 до 7 лет, описаны больные, жившие 30 лет. Возможны улучшение и даже выздоровление. Полное клиническое улучшение у больных говорит о принципиальной возможности обратного развития процесса.
К каким докторам следует обращаться если у Вас Болезнь Маркиафавы-Микели:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни Маркиафавы-Микели, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Дефицит кислой мальтазы. Болезнь Мак Ардла - дефицит миофосфорилазы.
Дефицит кислой мальтазы — это аутосомно-доминантное заболевание из группы болезней накопления гликогена. В основе патогенеза лежит дефицит лизосомальной альфа-гликозидазы, которая в норме участвует процессе превращения гликогена в глюкозу. В младенческом возрасте наблюдается генерализованный гликогенез с тяжелой кардиомиопатией, который носит название болезни Помпе. Более легкие формы болезни Помпе (с незначительным поражением сердца или без него) встречаются у взрослых.
В таких случаях отмечается медленно прогрессирующая миопатия с дыхательной недостаточностью. У детей с болезнью Помпе наблюдаются гипотония, макроглоссия, кардиомегалия и гепатомегалия. Обычно эти дети быстро погибают. На аутопсии обнаруживаются патологические изменения в мышцах диафрагмы, двуглавых мышцах плеча, мышцах плечевого пояса и аддукторах бедра. По данным электрофизиологических исследований выявляются миотонические изменения, хотя клинических проявлений миотонии нет. Вакуольная миопатия с высоким уровнем гликогена проявляется клинически.
Диагноз подтверждается исследованием кислой мальтазы в лейкоцитах или моче. Пренатальная диагностика вакуольной миопатии основана на данных биопсии ворсин хориона. В настоящее время методов специфической терапии не разработано. Больным можно рекомендовать дыхательную гимнастику.
Болезнь Мак Ардла (дефицит миофосфорилазы)
Болезнь Мак Ардла (дефицит миофосфорилазы) — это заболевание, встречающееся как у детей, так и у взрослых. Больные не переносят физические нагрузки — даже после минимальных упражнений у них появляются сильные миал-гии, патологическая усталость, напряжение мышц, миоглобинурия, почечная недостаточность, потеря массы тела. У некоторых наблюдаются эпилептические припадки. Отдельные пациенты приспосабливаются к своему заболеванию и избегают сильных кратковременных мышечных усилий, которые чаще всего вызывают обострение заболевания. Болезненные мышечные судороги и миоглобинурия обычно развиваются уже во взрослом возрасте.
Повышается уровень креатинкиназы. На ЭМГ отмечаются изменения, характерные для миотонии. Наложение жгута на предплечье (т. н. ишемическая нагрузка) не приводит к повышению уровня лактата в венозной крови. Заболевание наследуется аутосомно-доминантно. Ген, ответственный за выработку мышечной фосфорилазы, расположен в хромосоме 11. Прогноз у таких больных относительно благоприятный. При биопсии мышц обнаруживаются отложения гликогена под сарколеммой на периферии. Диагноз болезни Мак Ардла подтверждается данными гистохимического исследования на фосфорилазу — фермент не выявляется у больных.
Эффективного лечения не существует, но можно рекомендовать придерживаться диеты с низким содержанием аминокислот с разветвленными цепочками.
Недостаточность карнитин-О-пальмитилтрансферазы также клинически проявляется болезненными мышечными судорогами, миалгиями, миоглобинурией. Могут наблюдаться почечная и дыхательная недостаточность вследствие миоглобинурии. Провоцирующими факторами являются физические нагрузки. Способность больных к выполнению кратковременных, интенсивных упражнений не нарушена. Приступы заболевания обычно наблюдаются при длительных нагрузках. Рабдомиолиз может развиться, если больной голодает, длительно пребывает на холоде, употребляет в пищу много животных жиров, а также после вирусных инфекций или общей анестезии.
Заболевание наследуется аутосомно-доминантно. Дефектный ген локализован в хромосоме 1. Уровень креатинкиназы нормален. Специфическое лечение не разработано.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Течение миастении. Лечение миастении.
Для того чтобы назначить правильное лечение больному МГ, необходимо хорошо разобраться в особенностях течения заболевания. Кроме того, что очень трудно объяснить механизм развития заболевания, существует множество вариантов его клинического течения. Далее приведены некоторые общие наблюдения.
1. Приблизительно в 50% случаев заболевание манифестирует глазными симптомами, а у 80% больных эти признаки появляются в течение первого месяца болезни. У 10% больных МГ начинается со слабости бульбарной мускулатуры, у 10% - со слабости в конечностях, у 10% — с общей слабости, у 1% — со слабости дыхательной мускулатуры. На второй месяц заболевания у 40% пациентов наблюдаются изолированные глазные симптомы. У 40% симптомы генерализуются. В 10% случаев отмечаются симптомы, ограниченные конечностями и также в 10% случаев — бульбарной мускулатурой. Изолированная глазная форма заболевания на всем его протяжении наблюдается у 15—20% больных.
2. У большинства больных с начальными глазными симптомами в течение первого года заболевания развивается слабость в других мышечных группах (у 90% больных генерализованной формой МГ генерализация произошла в первые 12 месяцев болезни). Слабость достигает своего максимума в первые три года после начала заболевания. Смерть вследствие МГ — очень редкое явлением последние годы.
3. Спонтанная длительная ремиссия наблюдается у 10—15% пациентов, обычно в первые два года заболевания.
4. У большинства больных МГ отмечается прогрессирование клинических симптомов в первые 2—3 года заболевания. Однако, возможен и другой характер течения: как уже упоминалось выше, у 15—20% больных имеют место изолированные глазные симптомы, а у 10—15% развивается спонтанная ремиссия.

Лечение миастении
Ингибиторы холинэстеразы (ИХЭ) — безопасные и эффективные средства. Это препараты первого ряда для лечения больных с любой формой МГ.
Ингибиторы холинэстеразы снижают гидролиз ацетилхолина (АХ), усиливают накопление АХ в постсинаптической мембране никотиновых рецепторов. ИХЭ, применяемые при МГ, обратимо связываются с холинестеразой. Препараты не проходят через гематоэнцефалический барьер и обычно не вызывают побочных эффектов со стороны ЦНС. Всасывание из желудочно-кишечного тракта неполное и вариабельное — биодоступность препаратов при приеме внутрь составляет около 10%.
Мускариновые вегетативные побочные эффекты нередко наблюдаются при приеме ИХЭ. К ним относятся: боли в животе, диарея, саливация, слезотечение и при значительной выраженности брадикардия.
Холинергический криз. Опасным осложнением избыточного приема ИХЭ является развитие холинергического криза, проявляющегося слабостью скелетной мускулатуры. Большему риску этого осложнения подвержены больные, получающие ИХЭ парентерально. Крайне редко, даже у пациентов с выраженными мускариновыми холинергическими побочными эффектами (описаны единичные случаи), выраженная холинергическая слабость наблюдается в случае получения ИХЭ внутрь.
Наиболее распространенные ИХЭ перечислены в таблице.
- Пиридостигмин (местинон, калимин) — это наиболее часто применяемый препарат для длительной оральной терапии. После приема препарата эффект обычно начинается через 15—30 минут. Пик эффективности приходится на первый— второй час после приема, и действие постепенно угасает в течение последующих трех—четырех часов. Прием пиридостигмина обычно начинают с 30—60 мг два—три раза в сутки (в зависимости от симптоматики). Лучший эффект отмечается у большинства больных при использований 60 мг препарата каждые четыре часа. При увеличении доз повышается вероятность развития побочных мускариновых холинергических эффектов. Изредка больным требуется доза более 1000 мг/сут., которую они неплохо переносят при приеме препарата каждые 2— 3 ч. Пациенты, страдающие слабостью бульбарной мускулатуры, нередко стараются принимать препарат за 1 ч до еды, чтобы улучшить функции пережевывания пищи и глотания. Выраженность мускариновых побочных эффектов пиридостигмина минимальна по сравнению с другими ИХЭ.
- Кроме наиболее распространенной лекарственной формы пиридостигмина — таблеток 60 мг, существуют и другие формы препарата. Детям или больным с трудностями глотания обычно назначают сироп пиридостигмина. Пролонгированная форма — таблетки пиридостигмина 180 мг иногда применяются пациентами на ночь. Однако скорость высвобождения препарата и абсорбции предсказать трудно. Эти факторы ограничивают применение пролонгированной лекарственной формы.
- Парентеральное введение пиридостигмина может быть показано больным с тяжелой дисфагией или во время хирургических вмешательств. Доза пиридостигмина для внутривенного введения составляет 1/30 от энтеральной дозы.
- Неостигмин (прозерин) действует менее длительно, чем пиридостигмин и обладает более выраженными побочными мускариновыми эффектами.
- Амбенониум оказался одним из наиболее эффективных средств для лечения МГ. К его побочным эффектам относится головная боль.
- Тем больным, у которых наблюдаются выраженные мускариновые побочные эффекты при приеме ИХЭ в дозах, необходимых для лечения, рекомендован прием препаратов антихолинергического действия, например, атропина сульфат (0,4-0,5 мг внутрь) или гликопирролат (робинул) 1 мг для постоянного употребления или при каждом приеме ИХЭ.
- Больные с легкой формой заболевания обычно хорошо себя чувствуют при регулярном приеме ИХЭ. Однако пациентам с умеренной или тяжелой формами МГ обычно требуется более радикальная терапия.
Метаболическая миопатия
Метаболические миопатии - большая группа заболеваний, проявляющихся снижением толерантности к физической нагрузке вследствие нарушения обмена веществ в мышечной ткани или недостатка ферментов. Симптомами заболевания являются мышечная слабость, боли, судороги, парезы, трудность выполнения длительной физической нагрузки, формирование контрактур. Диагноз устанавливается на основании анамнеза, исследования содержания ферментов в кровяном русле, биопсии мышцы. Лечение включает в себя дието- и витаминотерапию, применение стероидов и ортопедической коррекции.
Общие сведения
Метаболические миопатии представляют собой обширную группу наследственных и приобретенных заболеваний, связанных с нарушением обмена веществ (гликогена, липидных фракций или АТФ) в мышцах. Патологический процесс проявляется как у детей, так и у взрослых. Наследственные формы склонны к прогрессированию патологического процесса. Актуальность метаболических миопатий связана с возможностью развития преходящих контрактур в ответ на невозможность мышечной ткани использовать энергический потенциал из глюкозы (болезнь Мак-Ардля) или липидов (недостаточность карнитина) с расщеплением АТФ. Метаболические миопатии развиваются примерно в 5 клинических случаях из 10000.
Причины развития метаболических миопатий
Основной причиной развития врожденных миопатий с нарушением метаболизма в мышечных тканях является наследственная предрасположенность (поэтому их еще называют семейными миопатиями); передача дефектного гена осуществляется аутосомно-рецессивно, реже доминантно или по материнской линии. Могут быть и случайные, или спорадические генные мутации. Патологический процесс при миопатиях такого рода может быть связан с патологией ионных каналов мышечных мембран (семейный периодический паралич), с нарушением функции митохондрий (вызванными мутациями в митохондриальном геноме). При гликогенозах наблюдается недостаточность ферментов, участвующих в обмене гликогена с последующим накоплением последнего в тканях, прежде всего в печени и скелетных мышцах. К примеру, при болезни Мак-Ардля патологический процесс связан с дефицитом фермента мышечной фосфорилазы, в результате чего нарушается процесс гликолиза, необходимый для обеспечения мышц энергией.
Приобретенные метаболические миопатии могут быть вызваны эндокринными заболеваниями (гипертиреоз, гипотиреоз, гиперальдостеронизм, болезни Кушинга и Аддисона, гиперфункция паращитовидных желез, акромегалия и прочие), быть следствием хронических интоксикаций (алкоголизм, ятрогенное воздействие и пр.); хронических заболеваний легких, почек (уремия, хроническая почечная недостаточность, электролитные нарушения), печени (печеночная недостаточность) и сердечно-сосудистой системы; синдрома мальабсорбции; дефицита витаминов группы D и E.
Классификация метаболических миопатий
Выделяют первичные (или врожденные) нарушения метаболизма мышечной ткани. К этой группе следует отнести заболевания, связанные с нарушением обмена гликогена: болезнь Помпе, болезнь Андерсена, болезнь Кори-Форбса, болезнь Мак-Ардля, болезнь Таури, дефицит киназы фосфорилазы b, дефицит ФГК, дефицит фосфоглицеромутазы, дефицит ЛДГ. К группе наследственных нарушений обмена липидов в мышцах относят: дефицит карнитина, недостаточность ацетил-КоА-дегидрогеназы и дефицит КПТ. Митохондриальными миопатиями являются: дефицит ферментов редуктазы, цитохрома b, b 1; дефицит АТФ. К нарушениям метаболизма пуринов относят: дефицит фермента МАДА (миоаденилдезаминазы).
Кроме этого, выделяют также и вторичные (приобретенные) метаболические миопатии, являющиеся следствием эндокринных болезней или других патологических состояний организма.
Симптомы метаболической миопатии
Нарушения обмена гликогена и глюкозы. При избыточном скоплении гликогена, пациенты достаточно хорошо чувствуют себя в покое, вне физической нагрузки. При повышении активности, недостаточность ферментов препятствует формированию энергии из углеводов. Обычно через несколько минут после начала физической работы больные начинают жаловаться на утомляемость, мышечную слабость. Если интенсивность нагрузки не уменьшить, может возникнуть мышечная контрактура, которая может перерасти в тяжелое поражение мышечной ткани.
При болезни Мак-Ардля возникают сильные мышечные боли, проксимальные мышечные гипотрофии, а также болезненное уплотнение и слабость мышц с последующим развитием контрактуры. Последняя обусловлена нарушением расслабления мышц, из-за дефицита энергии. При нагрузке может формироваться распад мышц с развитием миоглобинурии, явлениями тяжелой почечной недостаточности. Возможна слабость сердечной мускулатуры с развитием сердечной недостаточности.
Болезнь Андерсена и Кори-Форбса, приводят к тяжелым поражениям печени, сердца, контрактурам; нарушениям физического развития в детстве в сочетании с умеренным поражением мышц.
Дефицит мальтазы может развиваться в виде младенческой формы (сразу после рождения) с явлениями мышечной слабости и застойными явлениями в сердце. Болезнь прогрессирует и чаще всего приводит к летальному исходу; пациенты не доживают и до 2 лет. Вторая форма болезни начинает себя проявлять в раннем детском возрасте. Чаще всего связана с дыхательными нарушениями и выраженной слабостью мышц нижних конечностей. Взрослая форма болезни характеризуется безболезненной слабостью мышечного пояса верхних и нижних конечностей и дыхательной мускулатуры.
Нарушения липидного обмена. Большее число неврологических заболеваний связано с дефицитом липидных фракций. Некоторые из них могут проявляться только в снижении способности переносить физические нагрузки. Дефицит карнитина и фермента КПТ являются классическими примерами нарушения липидного обмена. Синдром дефицита карнитина проявляется прогрессирующей мышечной слабостью; поражаются мышцы конечностей, а также лицевые и глоточные мышцы. Синдром недостатка КПТ характеризуется приступами боли при физической нагрузке. Чаще всего болеют мужчины, у них может наблюдаться выраженная мышечная слабость и боль.
Митохондриальные миопатии. Поражаются мышцы конечностей, мышечные волокна глазного яблока (птоз, офтальмоплегия). Вовлекается в патологический процесс ЦНС (мозжечковая атаксия, эпилептические припадки, миоклонии, деменция и пр.), сердце (кардиомиопатии), почки (тубулопатия), печень, слуховой анализатор (нейросенсорная тугоухость). Обычно болеют дети и подростки, хотя патологический процесс может дебютировать и позже. Мышечная слабость либо имеет распространенный характер (повышенная утомляемость, плохая переносимость физических нагрузок), либо более ограниченный (локальное поражение мышц конечностей, лица или только наружных мышц глазного яблока). Характерна одышка при физической нагрузке, головные боли, напоминающие мигрень.
Диагностика метаболических миопатий
Постановка диагноза невозможна без сбора полной анамнестической картины и тщательного клинического обследования. Сложность связана с тем, что больные не предъявляют жалоб в состоянии покоя. Значимыми клиническими проявлениями являются: выраженные мышечные боли, слабость, судороги, изменения цвета мочи (миоглобинурия). Характерна симметричность поражения мышц. Также требуется тщательное неврологическое обследование на предмет выявления других клинических синдромов.
Назначается исследование ферментативной активности (кровь на ферменты КФК, АСТ, АЛТ, ЛДГ, альдолазы и пр.), ишемический тест на предплечье (позволяет выявлять гликогенозы). Электромиография или электронейрография позволяют выявить отклонения, которые характерны для миопатий. МРТ или КТ головного мозга назначаются с целью исключения органических изменений, которые также могут являться причиной мышечной слабости. По показаниям проводится томографическое исследование внутренних органов. Наиболее значимой в постановке диагноза является биопсия мышц с последующим морфологическим, гистохимическим и микроскопическим исследованием (позволяет поставить заключительный диагноз). Требуется консультация следующих специалистов: невролога, кардиолога, офтальмолога, нефролога; при проявлении метаболической миопатии в раннем детском возрасте - детского невролога, генетика и других.
Лечение метаболических миопатий
Лечение врожденных нарушений обмена веществ не представляется возможным. Применяется лишь симптоматическая помощь, которая выражается в диетотерапии с употреблением продуктов с высоким содержанием белка и приемом фруктозы. В сочетании с поливитаминами это может улучшать состояние больных при гликогенозах. При миоглобинурии вводят дополнительное количество жидкости для поддержания диуреза и профилактики почечной недостаточности. При митохондриальных миопатиях назначают лекарственные средства, улучшающие энергический метаболизм (L-карнитин, убихинон, рибофлавин, тиамин, витамины С и Е). Хорошие терапевтические результаты показывает применение глюкокортикостероидов. Показана также и ортопедическая коррекция.
При вторичных или приобретенных миопатиях лечение направлено на терапию основного заболевания: коррекцию эндокринных патологий, системных заболеваний, интоксикаций.
Прогноз и профилактика метаболических миопатий
Прогноз первичных миопатий достаточно неблагоприятный, особенно это касается младенческих форм и заболеваний, проявляющихся с первых недель жизни. Такие пациенты погибают в раннем детском возрасте. Более поздние или взрослые формы имеют более благоприятный прогноз. Течение и исход патологического процесса также зависит от вовлечения других органов и систем (сердца, печени, почек и пр.). Приобретенные метаболические миопатии имеют достаточно благоприятный прогноз, так как основные симптомы патологического процесса регрессируют после коррекции причинного заболевания и состояния.
Профилактика первичных метаболических миопатий заключается в медико-генетическом консультировании пары перед зачатием и в первом триместре беременности. Своевременное выявление и терапия вторичных миопатий позволяет избежать развития метаболических нарушений в мышечной ткани.
Гликогенозы у детей: болезни Гирке, Форбса-Кори, Андерсена, Мак-Ардла, Таруи
Гликогеноз I типа (болезнь Гирке) — не истинная миопатия, а заболевание, обусловленное дефицитом печеночного фермента глюкозо-6-фосфатазы, который в норме не присутствует в мышце, тем не менее у детей с этим заболеванием выявляется гипотония и умеренно выраженная мышечная слабость неизвестной этиологии. Гликогеноз II типа (болезнь Помпе) — аутосомно-рецессивно унаследованный дефицит гликолитического лизосомного фермента кислой мальтазы. Из 12 известных типов гликогенозов только II тип обусловлен дефектом лизосомного фермента. Аномальный ген картирован в локусе 17q23. Описано две формы заболевания.
Младенческая форма характеризуется тяжелой генерализованной миопатией и кардиомиопатией. У пациентов выявляется кардиомегалия, гепатомегалия, диффузная гипотония и мышечная слабость. Активность КФК в крови значительно повышена. Мышечная биопсия выявляет вакуолярную миопатию в сочетании с нарушением активности ферментов лизосом, таких как кислая и щелочная фосфатазы. Смерть обычно наступает в младенческом или раннем детском возрасте.
Поздняя детская или взрослая форма представлена миопатией с более легким течением, без увеличения сердца и печени. Клинические проявления могут отсутствовать до позднего детского или раннего зрелого возраста, однако возможно появление признаков мышечной слабости (обусловленной миопатией) и гипотонии даже в раннем младенческом возрасте. Активность КФК в крови значительно повышена, результаты мышечной биопсии имеют диагностическое значение даже на пресимптомной стадии заболевания.
Диагноз гликогеноза II типа подтверждается при количественном анализе активности кислой мальтазы при биопсии мышц или печени. При редком варианте дефицита кислой мальтазы с легким течением ее активность при биопсии мышц может находиться на нижней границе нормы с периодическим снижением до субнормального уровня, при этом результаты мышечной биопсии напоминают гликогеноз II типа, но изменения выражены более умеренно.
Другая форма — болезнь Данона — характеризуется Х-сцепленным рецессивным типом наследования, аномальный ген картирован в локусе Xql4. В основе заболевания лежит первичный дефицит протеина-2 мембран лизосом (LAMP2), который приводит к развитию гипертрофической кардиомиопатии, миопатии с поражением мышц проксимальных отделов конечностей и умственной отсталости.
Гликогеноз III типа (болезнь Форбса-Кори) обусловлен дефицитом фермента, расщепляющего гликоген (амило-1,6-глюкозидаза). Это наиболее распространенный гликогеноз с наименее тяжелыми клиническими проявлениями по сравнению с другими типами гликогенозов. В младенческом возрасте часто встречаются такие симптомы, как гипотония, мышечная слабость, гепатомегалия, гипогликемия при исследовании крови натощак, однако эти симптомы часто спонтанно исчезают и в детском возрасте, а также у взрослых клинические проявления могут отсутствовать.
В других случаях отмечается медленное прогрессирование атрофии мышц дистальных отделов конечностей, цирроза печени и сердечной недостаточности. При мышечной биопсии обнаруживаются минимально выраженные миопатические изменения, включающие вакуолизацию мышечных волокон.
Гликогеноз IV типа (болезнь Андерсена) обусловлен дефицитом фермента, участвующего в синтезе гликогена, приводящего к синтезу аномальных молекул гликогена — амилопектина — в печени, ретикулоэндотелиальных клетках, скелетной мускулатуре и сердечной мышце. Гипотония, генерализованная мышечная слабость, атрофия мышц и контрактуры — характерные признаки миопатического процесса. Большинство пациентов умирает до 4-летнего возраста в связи с развитием печеночной или сердечной недостаточности. Описаны отдельные случаи заболевания у детей без признаков нервно-мышечного заболевания.
Гликогеноз V типа (болезнь Мак-Ардла) обусловлен дефицитом мышечной фосфорилазы, наследуемым по аутосомно-рецессивному типу, аномальный ген картирован в локусе 1lql3. Основным клиническим проявлением заболевания служит непереносимость физической нагрузки, которая вызывает болезненный мышечный спазм (крампи), мышечную слабость и миоглобинурию; однако между приступами мышечная сила не снижена. Активность КФК в крови повышена только во время физической нагрузки. Характерным клиническим признаком служит отсутствие наблюдаемого в норме повышения уровня лактата в крови во время физической нагрузки, приводящей к ишемии.
Это обусловлено невозможностью превращения пирувата в лактат при анаэробных состояниях in vivo. Дефицит миофосфорилазы можно обнаружить с помощью гистохимических и биохимических методов в мышечном биоптате.
Редкая неонаталъная форма дефицита миофосфорилазы вызывает бульбарные расстройства в раннем младенческом возрасте, которые могут быть настолько выражены, что приводят к летальному исходу в периоде новорожденное™. В других случаях возможно медленное прогрессирование мышечной слабости, напоминающее мышечную дистрофию.
Отдаленный прогноз благоприятный. Пациенты должны научиться контролировать свой уровень физической нагрузки; тяжелой инвалидизации вследствие хронической миопатии или поражения сердца не отмечается.
Гликогеноз VII (болезнь Таруи) представляет собой дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V типа, оба заболевания характеризуются непереносимостью физической нагрузки, похожим клиническим течением и невозможностью превращения пирувата в лактат. Биохимическое исследование мышечных биоптатов позволяет дифференцировать эти два типа гликогенозов. Заболевание наследуется по аутосомно-рецессивному типу, аномальный ген картирован в локусе lcenq32.
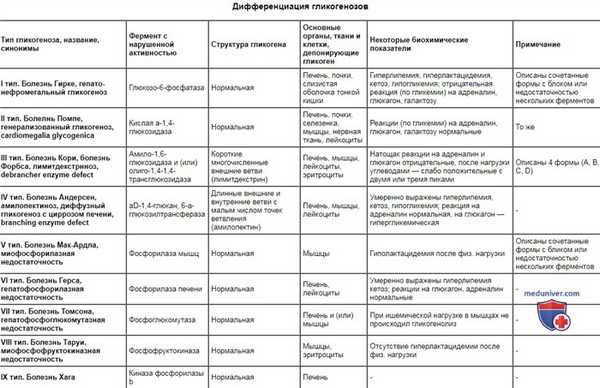
Дифференциация гликогенозов
| Тип гликогеноза, название, синонимы | Фермент с нарушенной активностью | Структура гликогена | Основные органы, ткани и клетки, депонирующие гликоген | Некоторые биохимические показатели | Примечание |
| I тип. Болезнь Гирке, гепато-нефромегальный гликогеноз | Глюкозо-6-фосфатаза | Нормальная | Печень, почки, слизистая оболочка тонкой кишки | Гиперлипемия, гиперлактацидемия, кетоз, гипогликемия; отрицательная реакция (по гликемии) на адреналин, глюкагон, галактозу | Описаны сочетанные формы с блоком или недостаточностью нескольких ферментов |
| II тип. Болелнь Помпе, генерализованный гликогеноз, cardiomegalia glycogenica | Кислая а-1,4-глюкозидаза | Нормальная | Печень, почки, селезенка, мышцы, нервная ткань, лейкоциты | Реакции (по гликемии) на адреналин, глюкагон, галактозу нормальные | То же |
| III тип. Болезнь Кори, болезнь Форбса, лимитдекстрнноз, debrancher enzyme defect | Амило-1,6-глюкозидаза и (или) олиго-1,4-1,4-трансглюкозидаза | Короткие многочисленные внешние ветви (лимитдекстрин) | Печень, мышцы, лейкоциты, эритроциты | Натощак реакции на адреналин и глюкагон отрицательные, после нагрузки углеводами — слабо положительные с двумя или тремя пиками | Описаны 4 формы (А, В, C, D) |
| IV тип. Болезнь Андерсен, амилопектиноз, диффузный гликогеноз с циррозом печени, branching enzyme defect | aD-1,4-глюкан, 6-а-глюкозилтрансфераза | Длинные внешние и внутренние ветви с малым числом точек ветвления (амилопектин) | Печень, мышцы, лейкоциты | Умеренно выражены гиперлипемия, кетоз, гипогликемия; реакция на адреналин нормальная, на глюкагон — гипергликемическая | - |
| V тип. Болезнь Мак-Ардла, миофосфорилазная недостаточность | Фосфорилаза мышц | Нормальная | Мышцы | Гиполактацидемия после физ. нагрузки | Описаны сочетанные формы с бликом или недостаточностью нескольких ферментов |
| VI тип. Болезнь Герса, гепатофосфорилазная недостаточность | Фосфорилаза печени | Нормальная | Печень, лейкоциты | Умеренно выражены гиперлипемия кетоз; реакции на глюкагон, адреналин нормальные | - |
| VII тип. Болезнь Томсона, гепатофосфоглюкомутазная недостаточность | Фосфоглюкомутаза | Нормальная | Печень и (или) мышцы | При ишемической нагрузке в мышцах не происходил гликогенолиз | - |
| VIII тип. Болезнь Таруи, миофосфофруктокиназная недостаточность | Фосфофруктокиназа | Нормальная | Мышцы, эритроциты | Отсутствие гиперлактацидемии после физ. нагрузки | - |
| IX тип. Болезнь Хага | Киназа фосфорилазы b | Нормальная | Печень | - | - |
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: