Дистрофия Лейдена
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Мне 24 года. Мой диагноз: прогрессирующая мышечная дистрофия. Первые симптомы появились лет в 7-8, стали слабеть ноги. В 14 лет стал замечать, что начали слабеть и руки. К врачу обратились в 16 лет. Меня сразу же положили в больницу; там мне объявили, что это заболевание не лечится. Так из цветущего, пышущего здоровьем молодого человека я постепенно стал превращаться в "ходячий скелет". Сейчас при росте 170 см я вешу 45 кг!
Вы в своей газете даете консультации по всевозможным заболеваниям, может быть, и мне вы поможете.
Сергей Б., Липецк
ПРОГРЕССИРУЮЩАЯ мышечная дистрофия - одно из самых тяжелых и широко распространенных наследственных заболеваний у человека, характеризующееся Х-сцепленным рецессивным типом наследования. Различают дистрофию Дюшенна, Беккера, Дрейфуса,
Ландузи-Дежерина, Лейдена-Мебиуса и ряд других форм заболеваний.
Наиболее распространенной (1 на 3500 новорожденных мальчиков) является мышечная дистрофия Дюшенна.
Она начинается в первые 1-3 года жизни обычно со слабости мышц тазового пояса. Уже на первом году жизни отмечается отставание в психомоторном развитии. Больные дети позднее начинают садиться, вставать, ходить. Постепенно развивается слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу утиной. Из горизонтального положения дети встают поэтапно с использованием рук (взбирание лесенкой). Отмечаются симметричные атрофии проксимальных групп мышц нижних конечностей (мышцы таза и бедра). Через 1-3 года атрофия распространяется на проксимальные группы мышц верхних конечностей. Атрофии мышц приводят к развитию у ребенка лордоза, крыловидных лопаток, осиной талии.
Вариант мышечной дистрофии Дюшенна - мышечная дистрофия Беккера - имеет более доброкачественное течение, но встречается почти в 10 раз реже (1 случай на 30 000 мужчин). Клинические проявления мышечной дистрофии Беккера обычно начинаются в 10-15 лет. При этом заболевании поражаются те же группы мышц, что и при дистрофии Дюшенна, но протекает оно мягче: начинается в более позднем возрасте, прогрессирует медленно; больные длительное время сохраняют работоспособность; сухожильные рефлексы долгое время остаются сохранными. Поражения внутренних органов менее выражены, интеллект сохранен. Больные нередко самостоятельно передвигаются даже в возрасте 50-70 лет.
По всей видимости, у молодого человека, написавшего нам письмо, наследственная прогрессирующая мышечная дистрофия Беккера. Однако точный диагноз может быть поставлен лишь лечащим невропатологом! Необходимо также наблюдение больного у хирурга-ортопеда, терапевта и профпатолога.
Мышечная дистрофия Дрейфуса характеризуется мышечной слабостью в ногах, затем в области плечевого пояса, лордозом, сколиозом, укорочением ахиллова сухожилия, ограничением подвижности шейного отдела позвоночника, умственной отсталостью. Начинается заболевание на первой декаде жизни.
Мышечная дистрофия Лейдена-Мебиуса (тазово-плечевая) - это дистрофия и слабость проксимальных групп мышц. Наиболее выраженные изменения захватывают тазовый и плечевой пояс.
Каковы причины прогрессирующих мышечных дистрофий?
Прогрессирующая мышечная дистрофия возникает в результате дефекта гена, кодирующего белок дистрофин. В мышцах перестает вырабатываться нормальный белок, и мышечная ткань постепенно замещается соединительной. В результате мышцы, в первую очередь конечностей, слабеют и атрофируются.
Заболевание значительно чаще наследуется мальчиками, чем девочками.
Профилактика прогрессирующих мышечных дистрофий заключается в генетическом консультировании.
Установить правильный диагноз и определить тип наследования врачам помогает наследственный анамнез, генеалогические анализы и лабораторные исследования.
Лечение
Несмотря на то, что поиски способов лечения ведутся во многих генных лабораториях мира, с сожалением приходится констатировать, что на сегодняшний день стопроцентных радикальных методов излечения прогрессирующих мышечных дистрофий нет. Возможна лишь поддерживающая терапия, с помощью которой удается улучшить состояние человека и продлить ему жизнь.
По определенной схеме больной должен систематически выполнять физические упражнения. Возможно использование протезов, позволяющих больным двигаться и замедляющих формирование сколиоза. При необходимости проводится поддержание дыхания.
Хирургическое вмешательство необходимо при наличии контрактур и фиксации суставов.
Эффективно замедляют прогрессирование заболевания и увеличивают мышечную силу у мальчиков специальные лекарственные препараты: глюкокортикоиды, витамины, животный белок.
Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни больного.
Люди, болеющие мышечными дистрофиями, должны соблюдать диету с достаточной энергетической ценностью, полноценным содержанием белков, жиров и углеводов.
Самый радикальный способ лечения прогрессирующих мышечных дистрофий во всем мире - это генотерапия (гены дистрофина и утрофина). Она заключается в том, что в каждую мышцу больного должен вводиться фрагмент гена, способного продуцировать нормальный мышечный белок. Генная терапия чрезвычайно перспективна и, по всей видимости, в ближайшие годы будет введена в медицинскую практику. Осталась нерешенной последняя задача - каким образом "нормальный" ген может попасть в организм человека.
Публикации в СМИ
К мышечным дистрофиям относят болезнь Дюшенна, Дрейфуса, Ландузи–Дежерина, Лейдена–Мёбиуса и ряд других заболеваний. Ниже приведена краткая характеристика наиболее распространённых мышечных дистрофий.
• Мышечная дистрофия Дюшенна (см. Дистрофия мышечная Дюшенна)
• Дистрофия плече-лопаточно-лицевая мышечная Ландузи–Дежерина (*158900, 4q35, ген FSHMD1A, Â ) — мышечная дистрофия с врождёнными дефектами отдельных мышц, например грудных, слабостью лицевой мускулатуры, мышц верхних конечностей, брюшной стенки, псевдогипертрофиями икроножных и дельтовидных мышц; на поздних стадиях появляется слабость мышц ног; часто сопровождается нейросенсорной тугоухостью, телеангиэктазиями сетчатки, образованием микроаневризм, экссудацией и кровоизлияниями в области жёлтого пятна, предсердной тахикардией, лёгочным сердцем; течение медленно прогрессирующее; частота — 0,9–2:100 000 населения. Синонимы • Плече-лопаточно-лицевая миопатия.
• Дистрофия мышечная тазово-плечевая Лейдена–Мёбиуса — дистрофия и слабость проксимальных групп мышц; наиболее выраженные изменения захватывают тазовый и плечевой пояс. Синонимы: • Лейдена дистрофия • Лейдена–Мёбиуса дистрофия.
• Дистрофия мышечная Эймери–Дрейфуса (синдром Эймери–Дрейфуса, *310300, Xq28, ген EMD, À ) — мышечная слабость в ногах, затем в области плечевого пояса, контрактуры, лордоз, сколиоз, укорочение ахиллова сухожилия, ограничение подвижности шейного отдела позвоночника, умственная отсталость, кардиомиопатия, АВ-блокада, изменения на ЭКГ; начало на первой декаде жизни; биопсия мышц — признаки первично-мышечного поражения (преимущественно быстрых мышечных волокон [типа II]).
• Мышечная дистрофия Фукуяма (см. Дистрофия мышечная Фукуяма)
Патоморфология • Для мышечных дистрофий характерен первично-мышечный тип поражения • Путём биопсии мышц выявляют дистрофию, фиброз, иногда воспалительные изменения • Иммуногистохимическое окрашивание в некоторых случаях позволяет выявить молекулярные дефекты, лежащие в основе некоторых мышечных дистрофий.
Генеалогический анализ и наследственный анамнез обычно позволяют установить тип наследования и предположить правильный диагноз.
Лабораторные исследования • Повышение уровня сывороточной КФК • Миопатические изменения на ЭМГ • Выявление сопутствующих нарушений — изменения на ЭКГ.
Лечение • Диета с достаточной энергетической ценностью, полноценным содержанием белков, жиров и углеводов • Специфическое лечение отсутствует • Поддерживающая терапия.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Код вставки на сайт
Дистрофии мышечные
К мышечным дистрофиям относят болезнь Дюшенна, Дрейфуса, Ландузи–Дежерина, Лейдена–Мёбиуса и ряд других заболеваний. Ниже приведена краткая характеристика наиболее распространённых мышечных дистрофий.
• Мышечная дистрофия Дюшенна (см. Дистрофия мышечная Дюшенна)
• Дистрофия плече-лопаточно-лицевая мышечная Ландузи–Дежерина (*158900, 4q35, ген FSHMD1A, Â ) — мышечная дистрофия с врождёнными дефектами отдельных мышц, например грудных, слабостью лицевой мускулатуры, мышц верхних конечностей, брюшной стенки, псевдогипертрофиями икроножных и дельтовидных мышц; на поздних стадиях появляется слабость мышц ног; часто сопровождается нейросенсорной тугоухостью, телеангиэктазиями сетчатки, образованием микроаневризм, экссудацией и кровоизлияниями в области жёлтого пятна, предсердной тахикардией, лёгочным сердцем; течение медленно прогрессирующее; частота — 0,9–2:100 000 населения. Синонимы • Плече-лопаточно-лицевая миопатия.
• Дистрофия мышечная тазово-плечевая Лейдена–Мёбиуса — дистрофия и слабость проксимальных групп мышц; наиболее выраженные изменения захватывают тазовый и плечевой пояс. Синонимы: • Лейдена дистрофия • Лейдена–Мёбиуса дистрофия.
• Дистрофия мышечная Эймери–Дрейфуса (синдром Эймери–Дрейфуса, *310300, Xq28, ген EMD, À ) — мышечная слабость в ногах, затем в области плечевого пояса, контрактуры, лордоз, сколиоз, укорочение ахиллова сухожилия, ограничение подвижности шейного отдела позвоночника, умственная отсталость, кардиомиопатия, АВ-блокада, изменения на ЭКГ; начало на первой декаде жизни; биопсия мышц — признаки первично-мышечного поражения (преимущественно быстрых мышечных волокон [типа II]).
• Мышечная дистрофия Фукуяма (см. Дистрофия мышечная Фукуяма)
Патоморфология • Для мышечных дистрофий характерен первично-мышечный тип поражения • Путём биопсии мышц выявляют дистрофию, фиброз, иногда воспалительные изменения • Иммуногистохимическое окрашивание в некоторых случаях позволяет выявить молекулярные дефекты, лежащие в основе некоторых мышечных дистрофий.
Генеалогический анализ и наследственный анамнез обычно позволяют установить тип наследования и предположить правильный диагноз.
Лабораторные исследования • Повышение уровня сывороточной КФК • Миопатические изменения на ЭМГ • Выявление сопутствующих нарушений — изменения на ЭКГ.
Лечение • Диета с достаточной энергетической ценностью, полноценным содержанием белков, жиров и углеводов • Специфическое лечение отсутствует • Поддерживающая терапия.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Дистрофия Лейдена
Конечностно-поясная мышечная дистрофия – вид мышечной дистрофии со множеством подтипов: некоторые из них являются аутосомно-рецессивными, а некоторые – аутосомно-доминантными. Время начала заболевания определяется типом наследования, но болезнь часто начинается в детстве. Симптомы медленно прогрессируют, поражая проксимальные мышцы. Диагноз ставится на основании анализа ДНК и биопсии мышц. Лечение направленно на поддержание функции и профилактику контрактур
Мышечные дистрофии являются наследственными прогрессирующими заболеваниями мышечной системы, возникающими из-за дефектов в одном или нескольких генах, необходимых для нормальной структуры мышц и их функционирования. Дистрофические изменения (например, некроз и регенерация мышечных волокон) видны на биоптатах.
Аналитические выводы, сделанные при помощи молекулярной биологии, заставили пересмотреть классификацию этих нарушений, и в данный момент номенклатура находится в переходной стадии. Аутосомно-доминантные формы были классифицированы как тазово-плечевая мышечная дистрофия (LGMD) 1A, -1B, -1C и так далее, а рецессивные формы были классифицированы как LGMD 2A, -2B, -2C и так далее. Тем не менее, после того, как буква Z была использована в 2016 году для обозначения рецессивной формы мышечной дистрофии конечностей LGMD, не осталось букв, чтобы назвать следующую форму, которая будет обнаружена. Отныне номенклатура будет начинаться с буквы D для доминантной и R для рецессивной формы, за которым следует число, указывающее сегмент, в котором определен локус, за которым следует белковый субстрат заболевания. Например, вариант заболевания, ранее называвшийся LGMD1F, теперь называется LGMD D2 транспортин 3.
Симптомы и признаки конечностно-поясной мышечной дистрофии
У пациентов, как правило, обнаруживается медленно прогрессирующая, симметричная слабость проксимальных мышц с или без вовлечения лицевых мышц и ослабленных или отсутствующих сухожильных рефлексов. В первую очередь может поражаться мускулатура таза или плечевого пояса. Симптомы при аутосомно-доминантных типах могут проявиться в любом возрасте, от раннего детского до взрослого. Развитие клинических симптомов при аутосомно-рецессивных типах начинается в детстве, и эти типы, в первую очередь, вызывают поражение тазового пояса.
Диагностика конечностно-поясной мышечной дистрофии
Анализ ДНК на наличие мутаций
В некоторых случаях показана биопсия мышц
Диагностика дистрофии Лейдена соответствует характерными клиническими данными, возрасту начала и семейному анамнезу и требует анализ мутаций ДНК из лейкоцитов периферической крови в качестве основного подтверждающего теста. Также можно провести гистологическое исследование мышц, иммуноцитохимию и вестерн-блоттинг.
Лечение конечностно-поясной мышечной дистрофии
Поддержание функции и профилактика контрактур
В процессе лечения тазово-плечевой дистрофии основное внимание уделяется поддержанию мышечной функции и предотвращению контрактур. Недавно изданное Американской академией неврологии руководство рекомендует проводить оценку сердечной деятельности пациентам с впервые диагностированной дистрофией Лейдена и высоким риском сердечно-сосудистых осложнений, даже при отсутствии у них сердечных симптомов. Пациенты с высоким риском дыхательной недостаточности должны пройти исследование функции легких. Все пациенты с тазово-плечевой мышечной дистрофией в идеале должны быть перенаправлены в многопрофильную клинику, которая квалифицируется на лечении нервно-мышечных расстройств.
Справочные материалы по лечению
1. Narayanaswami P, Weiss M, Selcen D, et al: Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 83:1453–1463, 2014. doi: 10.1212/WNL.0000000000000892
Дополнительная информация
Ниже следуют некоторые англоязычные ресурсы, которые могут быть информативными. Обратите внимание, что The manual не несет ответственности за содержание этих ресурсов.
Muscular Dystrophy Association: Информация об исследованиях, лечении, технологиях и поддержке пациентов с мышечной дистрофией
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Дистрофия Лейдена
ФГБУ Медико-генетический научный центр РАМН, Москва
Медико-генетический научный центр РАМН
Медико-генетический научный центр РАМН, Москва
Медико-генетический научный центр РАМН
Воронежская межобластная медико-генетическая консультация
Медико-генетический научный центр РАМН, Москва
Клинико-генетические характеристики поясно-конечностной мышечной дистрофии 2А типа
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2010;110(4): 79‑83
Дадали Е.Л., Щагина О.А., Рыжкова О.П., Руденская Г.Е., Федотов В.П., Поляков А.В. Клинико-генетические характеристики поясно-конечностной мышечной дистрофии 2А типа. Журнал неврологии и психиатрии им. С.С. Корсакова. 2010;110(4):79‑83.
Dadali EL, Shchagina OA, Ryzhkova OP, Rudenskaia GE, Fedotov VP, Poliakov AV. Clinical-genetic characteristics of limb girdle-muscular dystrophy type 2A. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2010;110(4):79‑83. (In Russ.).
ФГБУ Медико-генетический научный центр РАМН, Москва
Представлены результаты клинико-молекулярно-генетического обследования 26 больных из 24 неродственных семей с поясно-конечностной мышечной дистрофией (ПКМД) 2А типа, в возрасте от 12 до 60 лет. Длительность заболевания варьировала от 6 мес до 30 лет. Во всех случаях диагноз ПКМД 2А подтвержден молекулярно-генетическими методами на основании обнаружения мутации в гене CAPN3 в гомозиготном, компаунд-гетерозиготном или гетерозиготном состоянии. Наиболее частым фенотипом заболевания в российской популяции является вариант Лейдена-Мебиуса, характеризующийся первоочередным и преимущественным поражением мышц тазового пояса и голеней с постепенным распространением патологического процесса на мышцы плечевого пояса. Первым симптомом заболевания, на который указывают больные, являются ходьба на носках или трудности при подъеме по лестнице и беге. В противоположность ранее изложенным критериям Европейского нервно-мышечного центра, характерными симптомами заболевания являются рано возникающие контрактуры в голеностопных суставах, а также псевдогипертрофии икроножных мышц. Существует мажорная у российских больных мутация гена CAPN3 - c.550delA, которая обнаруживается в 70% случаев.
ФГБУ Медико-генетический научный центр РАМН, Москва
Медико-генетический научный центр РАМН
Медико-генетический научный центр РАМН, Москва
Медико-генетический научный центр РАМН
Воронежская межобластная медико-генетическая консультация
Медико-генетический научный центр РАМН, Москва
Поясно-конечностные прогрессирующие мышечные дистрофии (ПКМД) - группа клинически полиморфных и генетически гетерогенных заболеваний, характеризующихся преимущественным поражением мышц тазового и плечевого поясов. Частота всех ПКМД колеблется в различных популяциях от 5 до 70 больных на 1 млн населения. К настоящему времени описано 20 генетических вариантов этой группы заболеваний, которые подразделяются в зависимости от типа наследования на две подгруппы: ПКМД 1-го типа наследуются аутосомно-доминантно, ПКМД 2-го типа - аутосомно-рецессивно. В группе ПКМД 1-го типа идентифицировано 7 генетических вариантов, обозначаемых буквами английского алфавита (от А до G), в группе ПКМД 2-го типа - 13 вариантов (от А до M) [12]. Большая часть выявленных случаев ПКМД (до 85%) наследуется по аутосомно-рецессивному типу, их частота составляет в среднем 1:15 000 населения. Наиболее распространенным генетическим вариантом ПКМД с аутосомно-рецессивным типом наследования является 2A тип (OMIM: 253600), обусловленный мутациями в гене кальпаина 3 (CAPN3), на долю которого приходится 30-40% случаев ПКМД в большинстве европейских стран и до 80% среди высокоинбредной популяции басков Испании [22]. Интересно отметить, что одна из мутаций - c.550delA - наиболее часто выявляется у больных с ПКМД 2А из Хорватии, Болгарии, Словении, Турции, а также в ранее обследованной выборке российских больных [3, 5, 13, 14, 20]. В противоположность этому, при изучении выборки больных с ПКМД 2А из стран центральной Европы выявлена другая частая мутация - с.2362delAGinsTCATCT в экзоне 22, на долю которой приходилось более 30% всех мутаций в гене CAPN3 [11, 21].
Ген CAPN3 картирован на хромосоме 15q15.1-q21.1 [2]. Его продуктом является фермент семейства кальцийзависимых протеаз, основная функция которого заключается в обеспечении синхронизации процесса мышечного сокращения [16, 17, 20]. К настоящему времени описано более 150 мутаций в гене CAPN3 и их поиск продолжается [19, 22]. Показано, что в большинстве западноевропейских популяций наиболее часто мутации локализуются в 5, 11, 17, 21 и 22-м экзонах гена, при этом в отдельных популяциях или этнических группах обнаружены мажорные мутации, которые выявляются у большинства больных с ПКМД 2А [6, 7, 10, 18, 21, 22]. Идентификация таких мутаций, а также выявление особенностей клинических проявлений ПКМД 2А позволяют оптимизировать диагностический процесс и снизить затраты на проведение подтверждающей ДНК-диагностики [15].
Показано существование значительного меж- и внутрисемейного полиморфизма клинических проявлений ПКМД 2А. Так, по мнению C. Angelini и соавт.[1], существуют два основных клинических фенотипа ПКМД, обусловленных мутациями в гене CAPN3, отличающихся преимущественной локализацией патологического процесса и типом течения. Первый фенотип (форма Лейдена-Мебиуса) характеризуется преимущественным вовлечением в процесс мышц тазового пояса и бедер, второй (форма Эрба) - мышц плечевого пояса и проксимальных групп мышц верхних конечностей. По мнению авторов, в большинстве случаев ПКМД 2А первые признаки заболевания отмечаются в возрасте 12-18 лет. Наряду с этим, по данным международного консорциума по изучению нервно-мышечных заболеваний, существуют несколько фенотипов ПКМД 2А, различающихся возрастом начала и степенью генерализации процесса [4, 8, 9]. Первый вариант манифестирует в возрасте до 12 лет, характеризуется поражением мышц тазового и плечевого поясов, тяжелым течением и ранним возникновением контрактур в крупных суставах. Второй вариант, часто обозначаемый как мышечная дистрофия Лейдена-Мебиуса, манифестирует в возрастном интервале 13-29 лет и проявляется изолированным поражением мышц тазового пояса и бедер. При третьем варианте с началом в возрасте старше 30 лет отмечается поражение лишь мышц тазового пояса. Все эти клинические фенотипы являются аллельными генетическими вариантами, обусловленными мутациями в гене CAPN3.
Целью работы явилось описание клинико-генетических характеристик ПКМД 2А на основе изучения выборки больных, проживающих на территории России.
Материал и методы
Обследовали 26 больных из 24 неродственных семей с ПКМД 2А в возрасте от 12 до 60 лет. Длительность заболевания варьировала от 6 мес до 30 лет. Преобладали больные женского пола (18:9).
Диагноз заболевания ставили на основе клинического осмотра, генеалогических данных, электромиографического обследования, анализа уровня активности креатинфосфокиназы (КФК) в плазме крови и подтверждали результатами молекулярно-генетического анализа, направленного на поиск мутации в гене CAPN3.
Поиск мутаций в гене CAPN3 осуществляли методом прямого автоматического секвенирования по Сенгеру как с прямого, так и обратного праймеров, на приборе ABI Prism 3100 (Applied Biosystems) с использованием протокола фирмы производителя. В качестве матрицы для секвенирования использовали фрагменты ДНК, полученные после проведения полимеразной цепной реакции с использованием оригинальных олигонуклеотидных праймеров, которые синтезировались в НПО SYNTOL. Анализ результатов секвенирования осуществляли с помощью программ Chromas и BLAST.
Результаты и обсуждение
Во всех случаях диагноз ПКМД 2А был подтвержден молекулярно-генетическими методами, выявлены мутации гена CAPN3 в гомозиготном (одна и та же мутация обнаружена на обеих хромосомах), компаунд-гетерозиготном (на двух гомологичных хромосомах обнаружены разные мутации) или гетерозиготном (мутация обнаружена лишь на одной из пары гомологичных хромосом) состоянии.
Анализ выявленных у больных исследованной выборки мутаций в гене CAPN3, показал, что самой частой мутацией у российских больных, детектированной в 73% семей, является делеция одного нуклеотида с.550delA, приводящая к сдвигу рамки считывания. У 15% больных исследованной выборки эта мутация обнаружена в гомозиготном состоянии, у 58% - в компаунд-гетерозиготном состоянии, в сочетании с другими мутациями гена CAPN3, или гетерозиготном состоянии.
Три случая были семейными (больны два сибса), остальные - изолированными (большинство больных были единственными детьми в семьях). У всех больных наблюдался вариант Лейдена-Мебиуса, характеризующийся первоочередным вовлечением в процесс мышц тазового пояса и бедер с постепенным распространением мышечной слабости на мышцы плечевого пояса и туловища. Мышцы лица и глотки во всех случаях были интактны. Ни один из больных не имел фенотипов лопаточно-плечевой мышечной дистрофии Эрба или дистальной миопатии Миоши, которые в 10% случаев также могут быть обусловлены мутациями в гене CAPN3.
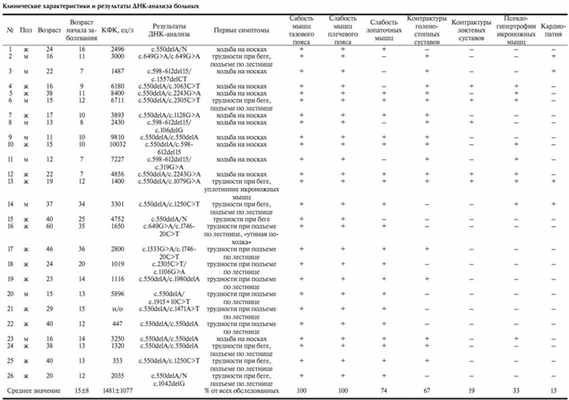
Клинические симптомы, выявленные у больных исследованной выборки, суммированы в таблице. Возраст манифестации заболевания варьировал от 7 до 36 лет (в среднем 15 лет), что соответствует ранее опубликованным данным. Приблизительно половина (12 из 27) больных в качестве первого симптома указывали появление ходьбы на носках, 14 - трудности при подъеме по лестнице и беге, и лишь 1 - уплотнение икроножных мышц. В клинической картине у всех обследованных доминировали симптомы слабости и гипотрофии мышц тазового и плечевого поясов. Вовлечение в процесс лопаточных мышц, проявлявшееся возникновением симптома «крыловидных лопаток», было отмечено только у 20 из 27 больных. Характерным признаком заболевания, выявленным у 67% больных, были контрактуры в голеностопных суставах, которые в ряде случаев достигали значительной степени выраженности, потребовавшей ахиллопластики. Наряду с этим у 5 (19%) больных контрактуры формировались и в локтевых суставах. У 33% больных были обнаружены псевдогипертрофии икроножных мышц, ранее считавшиеся нехарактерными для этого генетического варианта прогрессирующих мышечных дистрофий. Лишь у 4 больных с длительностью заболевания более 5 лет были выявлены признаки кардиомиопатии в виде нарушения метаболических процессов в миокарде, аритмии, тахикардии и блокады правой ножки пучка Гиса.
Анализ клинико-генетических характеристик больных с ПКМД 2А провели отдельно в 3 группах больных, выделенных в зависимости от возраста начала заболевания.
У половины (12 из 26) больных в возрасте от 11 до 22 лет с длительностью заболевания от 6 мес до 15 лет наблюдался первый вариант ПКМД 2А. Заболевание манифестировало в возрасте от 7 до 12 лет. У 11 из 13 больных этой группы первым симптомом заболевания, зафиксированным больными или их родителями, явилась тенденция ходьбы на носках. В 3 случаях первыми симптомами были трудности при подъеме по лестнице и беге, сопровождающиеся увеличением объема и уплотнением икроножных мышц. К моменту обследования все больные этой группы, кроме одной 22-летней девушки с длительностью заболевания более 15 лет, сохраняли способность к самостоятельной ходьбе и самообслуживанию. В клинической картине у всех больных этой группы доминировали симптомы вялого пареза с преимущественным поражением мышц тазового пояса. Вовлечение в патологический процесс мышц плечевого пояса происходило спустя несколько месяцев или лет от момента манифестации заболевания. У всех больных наблюдалось отсутствие или резкое угнетение сухожильных рефлексов с ног, причем первым выпадал ахиллов рефлекс. Сухожильные рефлексы с рук были снижены, но вызывались. Характерным клиническим признаком ПКМД 2А в обследованной выборке было раннее возникновение контрактур в голеностопных суставах, выявленные у всех больных. У 5 пациентов контрактуры обнаруживались и в локтевых суставах, а у 1 - в лучезапястных. Наличие контрактур в крупных суставах привело к необходимости проведения дифференциальной диагностики с прогрессирующей мышечной дистрофией (ПМД) Эмери-Дрейфуса с аутосомно-доминантным или Х-сцепленным рецессивным типом наследования. Однако этот диагноз был исключен при проведении ДНК-анализа, в результате которого не было обнаружено мутаций в генах ламина (LAMIN A/C) и эмерина (EMD). У 40% больных этой группы были выявлены псевдогипертрофии икроножных мышц, что давало основание в плане дифференциальной диагностики рассматривать другие варианты ПКМД, а также прогрессирующую мышечную дистрофию Дюшенна у больных мужского пола. Уровень КФК в этой группе больных варьировал от 1400 до 10 032 ед/л и снижался по мере прогрессирования заболевания.
Примером первого подтипа может служить история болезни пациентки Х., 16 лет (№4 в таблице).
Единственный ребенок в семье. Родилась от первой беременности, протекавшей без патологии, в срок, с массой 3000 г, длиной 50 см. Закричала сразу. Раннее моторное и психоречевое развитие протекало по возрасту. До 12 лет росла и развивалась без патологии. В 12-летнем возрасте при поведении планового биохимического обследования по поводу эрозивного гастродуоденита было обнаружено значительное повышение уровня АЛТ до 167 ед., АСТ до 233 ед. и КФК до 6180 ед/л в связи с чем поставлен диагноз «хронический гепатит неуточненной этиологии». При исследовании маркеров вирусных гепатитов патологии не выявлено. Ребенок предъявлял жалобы на повышенную мышечную утомляемость, трудности при беге и подъеме по лестнице. Выявлено изменение походки - стала ходить на носках. При осмотре больной в возрасте 16 лет выявлены умеренно выраженные признаки поясно-конечностной прогрессирующей мышечной дистрофии - гипотония, гипотрофия и снижение силы мышц тазового и плечевого поясов, гиперлордоз в поясничном отделе позвоночника. Мышцы лица, гортани и глотки не поражены. Походка «утиная», с упором на носки, при подъеме из положения на корточках использует приемы Говерса. Выявлена псевдогипертрофия икроножных мышц, а также тугоподвижность в голеностопных, локтевых и лучезапястных суставах. Сухожильные рефлексы с ног снижены, с рук без патологии. При проведении электромиографического исследования икроножной мышцы с использованием игольчатых электродов выявлены признаки первично-мышечного поражения, отмечено снижение амплитуды ПДЕ до 220 мкВ.
Таким образом, клинические проявления, повышение уровня КФК и первично-мышечный характер электромиографических показателей дали основание диагностировать прогрессирующую мышечную дистрофию. В связи с наличием псевдогипертрофий икроножных мышц, а также контрактур в крупных суставах было решено провести дифференциальную диагностику между ПМД Эмери-Дрейфуса, различными генетическими вариантами ПКМД и ПМД Дюшенна у носительницы мутации в гене дистрофина в гетерозиготном состоянии. Для исключения ПМД Дюшенна было проведено цитогенетическое исследование, направленное на выявление количественных и структурных перестроек хромосомы Х, а также ДНК-анализ для исключения несбалансированной лайонизации одной из Х-хромосом. При исследовании кариотипа больной патологии хромосомы Х не выявлено. Обнаружена структурная перестройка хромосомы 10, которая не могла привести к появлению симптомов ПКМД - 46,ХХ, inv (10)(p11.2-q21.1). Не было выявлено также несбалансированной лайонизации одной из Х-хромосом. Проведенные исследования позволили исключить ПМД Дюшенна. Аутосомно-доминантный вариант ПМД Эмери-Дрейфуса исключен на основании отсутствия мутаций в гене ламина А/С. Диагноз ПКМД 2А подтвержден на основании обнаружения двух мутаций (c.550delA, c.1063C>T) в гене CAPN3 в компаунд-гетерозиготном состоянии.
Вторая группа больных включала 11 человек с длительностью заболевания от 2 до 27 лет, с манифестацией в возрасте от 13 до 29 лет. У большинства (9 из 11) больных этой группы первым признаком заболевания была слабость в мышцах тазового пояса, проксимальных мышцах бедер, что проявлялось трудностями при подъеме по лестнице и из положения на корточках, беге и прыжках. Лишь два больных в качестве первых симптомов заболевания указывали на изменение походки в виде появления тенденции ходьбы на носках. Распространение патологического процесса также имело восходящий характер. Вовлечение в процесс мышц плечевого пояса наблюдалось спустя 2 года - 5 лет от момента манифестации процесса. Так же как и в первой группе больных, характерным признаком заболевания с более поздним началом были рано возникающие контрактуры в голеностопных суставах, выявленные у 40% больных. Однако ни у одного больного не были обнаружены контрактуры в локтевых и лучезапястных суставах, а нерезко выраженная псевдогипертрофия икроножных мышц отмечена лишь в 1 случае. Больные этой группы также длительно сохраняли способность к самостоятельной ходьбе. Только 1 больная в возрасте 40 лет стала пользоваться инвалидной коляской спустя 25 лет от момента манифестации заболевания. Показатели активности КФК в этой группе варьировали от 353 до 5896 ед/л. Как и у больных 1-й группы, отмечено снижение уровня КФК по мере прогрессирования заболевания. Так, минимальный уровень был обнаружен у больной, пользующейся инвалидной коляской, а максимальный - у больного с наименьшей длительностью заболевания.
Самой малочисленной оказалась третья группа больных, которая состояла лишь из 3 больных (2 женщины и 1 мужчина) в возрасте 37, 40 и 60 лет, с длительностью заболевания 3 года, 10 и 30 лет. Все больные самостоятельно передвигались и сохраняли способность к самообслуживанию. Первые признаки были отмечены больными в возрасте 34-36 лет в виде появления переваливающейся походки, трудности подъема по лестнице, из положения на корточках и при беге. Спустя 1-2 года появлялась слабость в мышцах плечевого пояса, трудности при подъеме рук выше горизонтального уровня, «крыловидные» лопатки, гипотрофия мышц и снижение их силы. Все больные использовали приемы Говерса. У 1 больного с длительностью заболевания около 3 лет отмечались умеренно выраженные псевдогипертрофии икроножных мышц, и у 1 больной - тугоподвижность в голеностопных суставах, возникшая спустя 8 лет от момента манифестации болезни. Уровень КФК варьировал от 3301 ед/л у больного с наименьшей длительностью заболевания до 1650 ед/л у больной с 30-летней длительностью болезни.
Таким образом, анализ клинико-генетических характеристик выборки российских больных с ПКМД 2А позволил сделать следующие выводы. Наиболее частым фенотипом заболевания является вариант Лейдена-Мебиуса, характеризующийся первоочередным и преимущественным поражением мышц тазового пояса и голеней с постепенным распространением патологического процесса на мышцы плечевого пояса. Первыми симптомами заболевания, на который указывают больные, являются ходьба на носках или трудности при подъеме по лестнице и беге. В противоположность ранее изложенным критериям Европейского нервно-мышечного центра, характерными симптомами заболевания являются рано возникающие контрактуры в голеностопных суставах, а также псевдогипертрофии икроножных мышц. Существует мажорная у российских больных мутация гена CAPN3 - c.550delA, которая обнаруживается в 70% случаев, следовательно, молекулярно-генетическая диагностика может начинаться с поиска этой мутации, что позволит существенно снизить затраты на ее проведение.
Дистрофия Лейдена
Мы занимаемся разработкой профессионального программного обеспечения, предназначенного для работы с медицинскими исследованиями.
Наша цель – сделать более удобной работу врачей-диагностов и клиницистов при работе с большими объемами данных, получаемых при исследованиях современным диагностическим оборудованием.
- Наши продукты – элементы для создания PACS (Picture Archiving Communication System) - полноценной системы получения, передачи, хранения и обмена медицинских исследований и медицинских изображений, обеспечения беспленочной технологии в лечебных учреждениях.
- Наши элементы PACS способны работать как в крупных лечебных учреждениях, с большим количеством разнородного медицинского диагностического оборудования, так и в небольших диагностических кабинетах, расширяя функциональные возможности единичных диагностических устройств.
- Основой наших программных продуктов является международный стандарт отображения, хранения и передачи медицинских данных, прежде всего медицинских изображений – DICOM .
- Наши программные продукты позволяют решать задачи, возникающие при построении сетей лечебных учреждений, предназначенных для обмена медицинской информацией, работать совместно с функционирующей или проектируемой медицинской информационной системой (МИС или HIS).
Проекты
Махаон DICOM Архив
Позволит вам создать единый расширяемый архив медицинских изображений, сохранять большие объемы данных, получаемых от разнообразного медицинского диагностического оборудования, обеспечить долгосрочное хранение медицинских исследований, объединить в единую сеть различные DICOM-устройства, создать сеть рабочих станций на базе Махаон Lite, обеспечить доступ к медицинским исследованиям, используя веб-интерфейс и многое другое.
Махаон Рабочая Станция
Позволит расширить диагностические возможности существующего медицинского оборудования и увеличить его пропускную способность, создав дополнительные рабочие места врачей, подключаемые к этому оборудованию, позволит осуществлять удаленное консультирование проведенных исследований, сравнение новых исследований с ранее проведенными, а также выполненными на других диагностических устройствах. Махаон Рабочая станция позволит вести локальный архив проведенных исследований на лазерных носителях с быстрым поиском проведенных ранее исследований. Махаон Рабочая Станция позволяет создать единую сеть с существующими DICOM-устройствами в лечебном учреждении. Махаон Рабочая Станция имеет специальные возможности для обработки изображений (MPR, DSA), а также модуль расширения функциональности для 3D- обработки и просмотра изображений.
Махаон Worklist сервер
Позволит обеспечить целостность хранения данных пациента в медицинской информационной системе (МИС), планирования медицинских исследований и передачи данных о пациентах и исследованиях медицинским устройствам.
Махаон Videograbber
Позволит получать изображения от нестандартных медицинских устройств и преобразовывать их в стандарт DICOM для последующей передачи их на другие DICOM-устройства и обеспечения их единообразного просмотра и анализа.
Махаон Net Lite
Позволит быстро и легко создать DICOM-сеть для обеспечения просмотра исследований, хранящихся в Архиве во всех врачебных кабинетах лечебного учреждения.
Махаон Медицинский справочник
Бесплатный справочник медицинской терминологии. Быстрый и удобный поиск по разделам. Онлайн обновления терминов с сервера.
Онлайн версия справочника
Махаон МКБ 10
Бесплатная электронная версия Международного классификатора болезней и проблем связанных со здоровьем 10-го пересмотра (МКБ 10).
Онлайн версия МКБ-10
4 Октября 2019 г.
Вышла версия 3.5
Поддержка систем и баз данных
Реализована поддержка систем Линукс программами Махаон Архив, Махаон Ворклист и Махаон Роутер. Обеспечено полное повторение функционала аналогичных продуктов Windows
Реализована поддержка баз данных Postgres SQL на Windows и Linux
Переписана анонимизация. Сделано полностью в соответствие стандарту. Анонимизируется около 300 тэгов. ФИО пациента анонимизируется в уникальное в пределах сессии работы программы, например: Anonymized 4RfdA45, у тэгов с удаленной информацией прописывается ‘Value removed’.
HL7 обрабатывает ескейп последовательности в полях E, F, R, S, T, X0D, X0A согласно стандарта. В обе стороны - и кодировка и чтение.
Улучшено прерывание запроса списков. Повторное нажатие приводит к немедленному прерыванию.
Визуальные элементы программ выполнены с учетом High DPI мониторов. Должны отображаться лучше на мониторах с высоким DPI.
Отображается текущая скорость выполнения всех сетевых операций а также число и объем обработанных элементов (файлов)
Рабочая Станция врача
Все оверлеи переписаны с GDI на GDI+. Заново написан движок вывода всех оверлеев
У всех оверлеев появилось сглаживание. Скорость отрисовки осталась практически на том же уровне
Ускорение построения тамбнейлов за счет создания миниатюр на добавлении в базу на видео и мультифреймах
Реализована обработка фильтров изображений в многопоточном режиме
Реализовано исправление данных в файлах (например - ФИО) в многопоточном режиме
Полноценное отображение видео в DICOM: открытие, перемотка, воспроизведение в окне в реальном времени, воспроизведение звука
Добавлена новая цветовая схема YBR_PARTIAL_420
В dicomdir дополнительно пишутся дата рождения пациента и пол
Вывод изображений и оверлеи
У всех оверлеев появилась возможность перетягивания надписей
Позиция надписи сохраняется в состоянии просмотра
Отображение относительного времени фрейма. Обрабатывается специальным тэгом в fieds.ovr
Отображение возраста пациента на момент исследование. Обрабатывается специальным тэгом в fieds.ovr
Появились специальные тэги - и - позиция текущего изображения в серии и размер серии
Режим w/l с которым открывается исследование может быть привязан к автоматическим установкам. Можно, например, открывать все CR как с полным дин. диапазоном, а всё остальное - как ‘из файла’.
Дописана конвертация в DICOM mpeg4 формат захваченных изображений. Захват звука и настройки входов звука и параметров также обрабатываются
Дописан импорт из AVI в DICOM mpeg4
При запросе данных из ворклиста будет учитываться набор символов, оператор исследования, рост и вес пациента и сохраняться в файлы.
Читайте также: