Доминантная краниометафизарная дисплазия
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Доминантная краниометафизарная дисплазия
Клинические данные. Данные осмотра. Несмотря на варьирующую экспрессивность гена, в большинстве случаев лицо имеет вполне характерный вид. Обычно на первом году жизни становится широким корень носа, крылья носа поднимаются, а носовые кости расширяются с обеих сторон по направлениям к слулам. Разрастающиеся гиперосто-ЗЫ носовых костей часто суживают носовые отверстия, что может привести к их полному закрытию. В результате этого у больных постоянно открыт рот.
Костная система. Скелетные аномалии обсуждены выше в разделе «Данные осмотра» и будут обсуждаться ниже в разделе «Лабораторные данные».
Нервная система. Хотя большинство больных, кроме нарушений слуха, неврологически здоровы, у некоторых из них могут наблюдаться периферический паралич лицевого нерва, головные боли или головокружения. Иногда отмечается снижение чувствительности на лице. Предполагают, что у этих больных в процесс вовлекается V пара череп-номозговых нервов (тройничный нерв). У некоторых из больных наблюдаются судорожные припадки или симптомы поражения пирамидных путей (Walker, Holt, Spranger et al., Rimoin et al., Kietzer, Paparella, Gladney, Monteleone, Cooper).
Орган слуха. Почти во всех случаях могут быть обнаружены различные степени глухоты. Это не редкий симптом болезни (Saunders). Потеря слуха начинается в детстве и медленно прогрессирует до умеренной или резко выраженной глухоты (от 30 до 90 дБ), развивающейся к третьему — четвертому десятилетию жизни. В 2 из 3 наблюдений SISI-тест, который вначале был отрицательным, к 12 годам жизни становился положительным (Gladney, Monteleone). Несмотря на то что глухота в основном проводящего типа, во многих случаях может наблюдаться и нейросенсорпый компонент (Saunders, Kietzer, Paparella, Gladney, Monteleone).
Вестибулярная система. Вестибулярные пробы нормальные (Paulsen et al.).
Лабораторные данные. Рентгенограммы. На рентгенограммах черепа обнаружены лобный и затылочный гиперостозы и/или склероз. Основание черепа обычно плотное с облитерированными параназальными синусами и с недостаточной пневматизацией сосцевидного отростка. Отмечается гипертелоризм. В течение первых 6 лет жизни метафизы длинных костей начинают приобретать булавовидную форму, выраженную значительно слабее, чем это наблюдается при болезни Пиле (метафизарной дисплазии), и могут быть на протяжении раннего детства минимальным и. У маленьких детей и в старшем возрасте встречается диафизарный склероз, но с возрастом он исчезает (Beckman, Walker). В коротких трубчатых костях обнаруживаются те же самые изменения, что и в длинных костях. Может быть расширена грудинная половина ключиц. Таз и латеральная часть позвоночника нормальные. В некоторых случаях отмечается сужение внутреннего слухового прохода (Rimoin et al.). Политомограммы показали, что область улитки заполнена костью (Gladney, Monteleone).
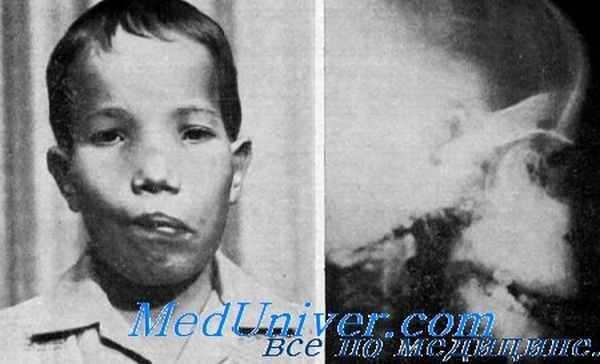
Патология. Возможно, скошенность метафизов, утолщение костей черепа и лица возникают вследствие недостаточной резорбции вторичной спонгиозной ткани. Сужение отверстий черепно-мозговых нервов, возникающее, вероятно, в связи с заполнением их костью, ведет к глухоте, а в некоторых случаях к потере зрения, нистагму, параличу лицевого нерва. Глухота является смешанной. Сужение внутреннего слухового прохода может быть установлено на рентгенограмме. Нейросенсорпый компонент глухоты, по-видимому, возникает в связи со сдавлением слухового нерва (Miller et al.). Причиной нарушения функции проводящего компонента в большинстве случаев, очевидно, является неподвижность основания стремени пли костные разращения на слуховых косточках, ведущие к их иммобилизации (Kietzer, Paparella).
Наследственность. Выявление заболевания в нескольких следующих друг за другом поколениях, так же как и передача болезни от мужчины к мужчине, отчетливо демонстрируют аутосомно-доминантное наследование (Brown, Harper, Spranger et al., Rimoin et al.).
Диагноз. Гиперостоз черепа наблюдается также при рецессивной краниометафизарной дисплазии, краниодиафизарной дисплазии, врожденной гинерфосфатазии, синдроме Шварца—Лелека, болезни Педжета, остеопетрозе, болезни Ван Бухема, фронто-метафизарной дисплазии, дизостеосклерозе и склеростеозе. Патология длинных костей при болезни Пиле (метафизарной дисплазии) может быть отграничена от того, что наблюдается при краниометафизарной дисплазии (Gorlin et al.). Преходящий дпафизарный склероз, наблюдающийся при краниометафизарной дисплазии в детстве, может выглядеть подобно тому, что отмечается при болезни Энгельманна.
Скошенные метафизы длинных костей могут быть также обнаружены у больных остеопетрозом, гиперплазией костного мозга, инфильтрацией костного мозга, как при болезни Гоше или лейкемиях, при некоторых дефицитных состояниях, таких, как излечивающаяся цыпга, или излечивающийся рахит, а также при некоторых токсических состояниях, таких, как свинцовое отравление. Однако пи при одном из этих состояний не обнаружен гиперостоз костей черепа.
Лечение. Слуховые аппараты могут уменьшить потерю слуха. Лицам с преимущественно проводящей глухотой нужно рекомендовать стапедэктомию.
Прогноз. Слух снижается вскоре после рождения и дефект слуха в дальнейшем может обнаружить некоторое прогрессировать, в большинстве случаев до подросткового возраста. Такие же различия в степени выраженности и прогресснрования наблюдаются при потере зрения и параличе лицевого нерва.
Выводы. Характеристика этого синдрома включает: 1) аутосомно-доминантное наследование; 2) склероз костей черепа с сужением отверстий черепно-мозговых нервов, в результате чего в некоторых случаях развиваются параличи черепно-мозговых нервов; 3) метафизариую дисплазию, поражающую длинные кости; 4) смешанную глухоту примерно в половине случаев.
Краниометафизарная дисплазия
Краниометафизарная дисплазия - остеохондродисплазия, характеризующаяся гиперостозом и склерозом черепно-лицевых костей и расширением метафизов. Часто данное заболевание принимают за болезнь Пайла (множественную метафизарную дисплазию), однако при краниометафизарной дисплазии наблюдается более выраженный гиперостоз черепа и меньшее расширение метафизов. Для заболевания характерно утолщение свода и склероз основания черепа, утолщение костей лицевого черепа, макроцефалия, относительно короткий нос, отличительным признаком является толстый костный выступ над переносицей. У пациентов наблюдается гипертелоризм глаз, экзофтальм, сдавление черепных нервов, головная боль, узкие носовые ходы и заложенный нос. В конечностях отмечается легкое или умеренное расширение и склероз метафизов, наиболее заметные на дистальном конце бедренной кости, Х-образное искривление ног.
Описаны две формы краниометафизарной дисплазии: с аутосомно-доминантным и аутосомно-рецессивным типом наследования. Аутосомно-доминантная форма болезни протекает довольно легко, и с возрастом характерные черты лица сглаживаются, симптомы обычно сводятся к последствиям сдавления черепных нервов (особенно лицевого и преддверно-улиткового), а иногда ограничиваются только склерозом черепных швов. При аутосомно-рецессивной форме, напротив, характерные черты лица с возрастом усиливаются, ухудшается зрение, нарастает тугоухость, нарушается прикус, развивается парез мимических мышц, иногда возникает атаксия, обратимая после декомпрессии задней мозговой ямки.
Наиболее часто встречается аутосомно-доминантная форма черепно-метафизарной дисплазии. К развитию данного заболевания приводят мутации в гене ANKH, локализованном на хромосоме 5p15.2, гомологичном мышиному гену прогрессирующего анкилоза. У мышей данный ген кодирует мембранный переносчик пирофосфата – главного ингибитора обызвествления и резорбции костной ткани. Специфические мутации в гене ANKH приводят также к семейному хондрокальцинозу (псевдоподагре).
Аутосомно-рецессивная краниометафизарная дисплазия встречается крайне редко. В литературе описаны несколько семейных и спорадических случаев заболевания из Бразилии, Португалии и Индии, большинство из которых наблюдались в близкородственных браках. Все известные случаи обусловлены уникальной миссенс-мутацией c.716G>A (p.Arg239Gln) в гене GJA1, выявленной у пациентов в гомозиготном состоянии. Ген GJA1 кодирует коннексин 43, и другие мутации в данном гене вызывают формирование глазо-зубо-пальцевой дисплазии (окуло-денто-дигитального синдрома) или изолированной синдактилии III типа.
В Центре Молекулярной Генетики для молекулярно-генетической диагностики аутосомно-доминантной краниометафизарной дисплазии проводится поиск мутаций в гене ANKH методом прямого секвенирования кодирующей последовательности и областей экзон-интронных соединений гена ANKH, для диагностики аутосомно-рецессивной формы краниометафизарной дисплазии проводится поиск мутаций в гене GJA1 методом секвенирования кодирующей последовательности данного гена.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Краниотубулярные дисплазии
Остеопетроз является наследственным заболеванием, характеризуется повышенной плотностью костной ткани и формированием аномального скелета.
Краниометафизарная дисплазия
Это аутосомно-доминантное заболевание Аутосомно-доминантные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения человека обусловлено мутациями гена ANKH. В раннем детстве образуются параназальные бугры, а также прогрессирующее расширение и утолщение костей черепа и нижней челюсти, что приводит к деформации лица и челюсти. Поражение костей вовлекает черепные нервы, вызывая дисфункцию. Исправление прикуса зубов Неправильный прикус Неправильный прикус является ненормальным контактом между зубами верхней и нижней челюсти. (См. также Обследование пациентов со стоматологическими заболеваниями [Evaluation of the Dental Patient]. Прочитайте дополнительные сведенияДиагноз краниометафизарной дисплазии предполагается при наличии типичных черепно-лицевых аномалий, которые иногда сочетаются с повышенной восприимчивостью к респираторным инфекциям, или нарушение может быть обнаружено во время оценки дисфункции черепных нервов, которые могут возникнуть в результате защемления у основания черепа. Как правило, проводят обычное рентгенологическое исследование. Рентгенологические изменения являются возрастными и обычно проявляются в возрасте 5 лет. Склероз является главной особенностью черепа. В трубчатых костях отмечаются булавовидные расширения метафизов, особенно в области дистальной части бедренной кости. Однако, эти изменения являются менее серьезными, чем при болезни Пайла Метафизарная дисплазия (болезнь Пайла) Краниотубулярные дисплазии включают минимальный остеосклероз с нормальным формированием скелета. Остеопетроз является наследственным заболеванием, характеризуется повышенной плотностью костной. Прочитайте дополнительные сведения . Позвоночник и таз не поражены.
Лечение кранио-метафизарной дисплазии состоит из хирургической декомпрессии затронутых нервов и ремоделирования тяжелых костных аномалий. Тем не менее, возобновление роста всё-таки происходит.
Фронтометафизарная дисплазия
Это расстройство становится очевидным в раннем детстве. Надглазничный валик выдается, напоминая забрало (шлем) рыцаря. Нижняя челюсть гипопластичная с передним сужением; аномалии зубов являются зарактерными. Глухота развивается в зрелом возрасте, потому что склероз сужает внутреннее слуховое отверстие и среднее ухо, или может привести к деформации слуховых косточек. Длинные кости ног умеренно изогнуты. Прогрессирующие контрактуры конечных фаланг пальцев могут имитировать артрит. Рост и общее состояние здоровья в норме.
Диагноз фронтометафизарной дисплазии подозревают при потере слуха у пациентов с признаками скелетных аномалий, описанных выше. Как правило, проводят обычное рентгенологическое исследование. На рентгене видны костные разрастания лобной области; пятнистый склероз наблюдается в своде черепа. Тела позвонков диспластические, но не склеротические. Подвздошные гребни резко расширены, и вход в таз деформирован. Эпифизы бедренной кости со стороны головки уплощены с расширением головок бедренных костей и дисплазией тазобедренного сустава (деформации бедра). Кости пальцев несформированы, имеют эрозию и сужение суставной щели.
Корректирующее хирургическое лечение показано при сильно уродующих деформациях, в том числе, тяжелой микрогнатии Микрогнатия (маленькая нижняя челюсть) При рождении челюсти могут отсутствовать, быть деформированными или не полностью развитыми. (См.также Введение в врожденные черепно-лицевые и скелетно-мышечные расстройства (Introduction to. Прочитайте дополнительные сведенияМетафизарная дисплазия (болезнь Пайла)
Это редкое аутосомно-рецессивное заболевание Аутосомно-рецессивные Генетические нарушения, вызванные изменениями в одном гене («Менделевские нарушения»), являются самыми простыми для анализа и наиболее хорошо поняты. Если экспрессия признака требует только. Прочитайте дополнительные сведения вызвано деффектами гена SFRP4. Его часто путают с краниометафизарной дисплазией Краниометафизарная дисплазия Краниотубулярные дисплазии включают минимальный остеосклероз с нормальным формированием скелета. Остеопетроз является наследственным заболеванием, характеризуется повышенной плотностью костной. Прочитайте дополнительные сведения . Пострадавшие люди являются клинически нормальными, за исключением наружного отклонения голени Варусное колено (О-образное искривление ног) и наружное отклонение голени(Х-образные ноги) Два основных типа коленных или бедренно-большеберцовых угловых деформаций – варусное колено (саблевидная нога) и наружное отклонение голени (вывернутые внутрь колени). При отсутствии лечения. Прочитайте дополнительные сведения и хрупкость костей.
Диагноз метафизарной дисплазии обычно ставят при проведении рентгенографии по несвязанным причинам. Рентгенологические изменения являются патогномоничными. Длинные кости недостаточно деформированы, и наружный слой кости, как правило, тонкий. Трубчатые кости ног имеют грубое расширение книзу, подобно колбе Эрленмейера, особенно в дистальной части бедренной кости. Кости малого таза и грудной клетки расширены. Тем не менее, череп, в основном, не затронут.
Лечение метафизарной остеодисплазии часто не является необходимым, но может включать методы ортодонтического лечения при дентальных пороках развития, или ортопедической хирургии для клинически значимых деформаций скелета.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Краниометафизарная дисплазия
Краниометафизарная дисплазия – наследственное состояние из группы остеохондродисплазий, характеризующееся аномалиями развития черепа и метафизов костей конечностей. Симптомами данного заболевания являются гипертелоризм, пороки развития лица, нередко уродующие больного, аномальное формирование носовых ходов с нарушением их проходимости, иногда головные боли и искривления конечностей. Диагностика краниометафизарной дисплазии осуществляется на основании изучения настоящего статуса пациента, рентгенологических данных и результатов молекулярно-генетических исследований. Специфического лечения данной патологии не существует, используют симптоматическую терапию, в некоторых случаях проводят хирургические вмешательства для облегчения дыхания пациента и улучшения его внешнего вида.
Общие сведения
Краниометафизарная дисплазия – группа наследственных заболеваний, которые приводят к порокам развития костей лицевого и мозгового отделов черепа различной степени выраженности в сочетании с аномалией метафизов длинных трубчатых костей и другими нарушениями. Ранее это состояние относили в одну группу с болезнью Пайла (множественная метафизарная дисплазия, краниометафизарная дисплазия Пайла), однако в настоящее время его выделяют в отдельную нозологическую единицу. Это связано с тем, что при данном заболевании превалирующими являются именно аномалии строения черепа (деформации, гиперостозы и склерозы костей), тогда как патологии иных отделов скелета выражены слабо. Существуют две основные формы краниометафизарной дисплазии, отличающиеся между собой механизмом наследования (аутосомно-доминантный и аутосомно-рецессивный типы), клиническими проявлениями и выраженностью нарушений. За счет наследования посредством аутосом патология с равной долей вероятности поражает как мужчин, так и женщин. Встречаемость краниометафизарной дисплазии точно не установлена, доминантный тип регистрируется во много раз чаще рецессивного.
Причины краниометафизарной дисплазии
Основной причиной более частой, но более легкой аутосомно-доминантной формы краниометафизарной дисплазии становятся мутации в гене ANKH, располагающемся на 5-й хромосоме. Ген кодирует белок, который является мембранным переносчиком пирофосфата, участвующего в угнетении процессов обызвествления костной ткани и ее резорбции. В результате генетического дефекта у белка-переносчика изменяется структура, и он становится неспособным полноценно выполнять свои функции, что приводит к краниометафизарной дисплазии. На клеточном уровне это проявляется аномальным изменением активности остеокластов, развитием склероза и гиперостоза главным образом костей черепа. Также может нарушаться конфигурация основных отверстий основания черепа, что становится причиной компрессии некоторых сосудов и нервов и во многом определяет остальные симптомы краниометафизарной дисплазии (нарушения слуха, головные боли, поражения тройничного и лицевого нервов). В генетике известны и другие заболевания, обусловленные поражением гена ANKH, в частности – наследственная псевдоподагра или семейный хондрокальциноз.
Намного более редкая аутосомно-рецессивная форма краниометафизарной дисплазии обусловлена дефектом гена GJA1, локализованного на 7-й хромосоме. Продуктом его экспрессии является белок под названием коннексин 43, принимающий активное участие в формировании межклеточных (межщелевых) контактов во многих тканях, что позволяет клеткам обмениваться низкомолекулярными соединениями. Патогенез развития краниометафизарной дисплазии при миссенс-мутации c716G>A неизвестен, основная проблема сводится к выявлению причин изолированного поражения костей черепа и метафизов костей при относительном отсутствии патологий других органов. Учитывая, что наибольшее количество коннексина 43 у человека находится в сердечной ткани, вопрос отсутствия патологий сердца при данной мутации на сегодняшний день представляет собой загадку для большинства врачей-генетиков. Так же, как и в случае аутосомно-доминантной формы краниометафизарной дисплазии, при этом типе заболевания наблюдаются многочисленные вторичные нарушения, обусловленные воздействием измененных костей и отверстий на нервные структуры.
Симптомы краниометафизарной дисплазии
Аутосомно-доминантный тип краниометафизарной дисплазии характеризуется более легким течением, нередко при рождении ребенка никаких симптомов заболевания не выявляется. Лишь к первому году жизни возникает гипертелоризм с расширением переносицы вследствие гиперостоза носовых костей. В дальнейшем краниометафизарная дисплазия приводит к сужению носовых ходов и нарушению их проходимости, поэтому у больных нередко приоткрыт рот из-за нарушенного носового дыхания. К 6-7 годам может начать определяться увеличение метафизов длинных трубчатых костей, что внешне проявляется увеличением размеров коленных и локтевых суставов. Примерно у половины больных краниометафизарной дисплазией развиваются нарушения слуха различной выраженности вплоть до полной глухоты – чаще всего это обусловлено компрессией слухового нерва в костном канале. Из-за сдавления нервов возникают и другие неврологические нарушения, возможны расстройства чувствительности на лице, невралгия тройничного нерва и головные боли.
Краниометафизарная дисплазия аутосомно-рецессивного типа характеризуется намного более выраженными пороками развития костей черепа и конечностей, признаки патологии часто выявляются сразу после рождения ребенка. У больных присутствует выраженный гипертелоризм, черты лица зачастую крайне несимметричны, деформации переносицы и носа могут приобретать признаки уродства. В ряде случаев наблюдаются макроцефалия, нижнечелюстной прогнатизм, другие нарушения прикуса и расположения зубов. По мере роста больного аномалии костей черепа могут усугубляться. Метафизы костей конечностей резко расширены, что нередко обуславливает вторичные деформации (например, Х-образное искривление ног). Как и в предыдущем варианте, при этой форме краниометафизарной дисплазии часто возникают разнообразные неврологические нарушения, вызванные сдавлением и травматизацией черепно-мозговых нервов. Они могут проявляться глухотой, нарушениями зрения, расстройствами кожной чувствительности на лице, парезом мимической мускулатуры и головными болями. Существуют отдельные описания больных, одновременно страдающих краниометафизарной дисплазией и умственной отсталостью, однако достоверных данных о взаимосвязи этих двух состояний на сегодняшний день нет.
Диагностика и лечение краниометафизарной дисплазии
Диагностика краниометафизарной дисплазии основывается на данных осмотра пациента, изучении его наследственного анамнеза, результатах рентгенологических исследований и молекулярно-генетических анализов. При осмотре определяются различные по выраженности аномалии развития костей черепа, что отражается на чертах лица больного. При этом аутосомно-доминантная форма, особенно у маленьких детей, довольно слабо проявляет себя в отношении этого симптома. При обоих типах краниометафизарной дисплазии практически всегда присутствуют гипертелоризм, расширение переносицы за счет разрастания носовых костей по направлению к скулам, сужение или непроходимость носовых ходов. В рамках физикального осмотра и дополнительных исследований могут быть также выявлены неврологические нарушения: снижение или отсутствие слуха, снижение зрения, симптомы со стороны тройничного и лицевого нервов.
Намного больше информации при краниометафизарной дисплазии дают рентгенологические методы исследования скелета. На рентгенограммах черепа при аутосомно-доминантной форме заболевания определяются уплотнение костной ткани в области затылочной кости, склероз основания черепа, пониженная пневматизация синусов и ячеек височной кости. В ряде случаев может выявляться склероз межкостных швов и расширение метафизов трубчатых костей. При аутосомно-рецессивном типе краниометафизарной дисплазии на рентгенограммах обнаруживаются схожие, но намного более выраженные нарушения, например – полное отсутствие околоносовых пазух, резкое сужение и иногда заполнение костной тканью отверстий черепно-мозговых нервов. Кроме того, наблюдается склероз не только основания, но и свода черепа, в ряде случаев определяются значительные деформации костей лицевого отдела. Немного сильнее, чем при доминантном типе, выражены расширение и склероз метафизов трубчатых костей.
Изучение наследственного анамнеза и генетическая диагностика также активно используются для определения краниометафизарной дисплазии. При этом можно сначала установить тип наследования заболевания, что позволяет скорректировать молекулярно-генетический анализ для поиска мутаций в конкретном гене. Для этого применяют метод прямого секвенирования генов ANKH и GJA1.
Специфического лечения краниометафизарной дисплазии не существует, назначают симптоматическую терапию с привлечением разнообразных хирургических техник. Последние, в частности, могут улучшить проходимость носовых ходов для облегчения дыхания. Уменьшить выраженность неврологической симптоматики можно путем расширения отверстий соответствующих черепно-мозговых нервов. Кроме того, пластические хирурги способны минимизировать выраженность эстетических дефектов при краниометафизарной дисплазии. Для улучшения слуха используют слуховые аппараты.
Прогноз и профилактика краниометафизарной дисплазии
Во многих случаях прогноз краниометафизарной дисплазии аутосомно-доминантного типа относительно выживаемости больного благоприятный – ни аномалии черепа, ни вторичные неврологические нарушения не приводят к тяжелым последствиям. Возникающие в детстве патологии слуха или зрения могут медленно прогрессировать до 20-30 лет, после этого ухудшений обычно не происходит. Аутосомно-рецессивный тип краниометафизарной дисплазии характеризуется более неблагоприятным прогнозом, поскольку костные нарушения черепа при этом состоянии имеют тенденцию к усугублению и могут с каждым годом все сильнее травмировать нервы. В некоторых случаях это приводит к параличам и расстройствам со стороны вегетативной нервной системы. Профилактика краниометафизарной дисплазии не разработана, для уменьшения риска развития осложнений при наличии такого состояния необходимо проводить регулярное обследование у невролога.
Метафизарная дисплазия

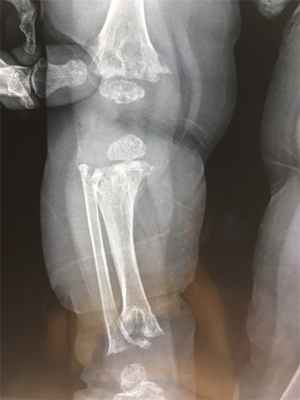
Метафизарная дисплазия – тяжелое и редкое заболевание костной ткани. У пациентов наблюдаются нарушения в строении стволов длинных костей и их метафизов, формирование широкого основания. По мере развития форма начинает напоминать лодочное весло, увеличивается губчатый внутренний слой. Пациенты с подобной патологией часто сталкиваются с деформацией скелета, патологическими переломами и другими осложнениями, угрожающими жизни и здоровью.
Общие сведения о болезни
Метафизарная дисплазия плохо изучена врачами, но доказана ее наследственная передача. Болезнь приводит к порокам развития костной ткани скелета, при которой наблюдается аномальное строение метафиза в длинных трубчатых костях. В зависимости от симптомов, тяжести поражения опорно-двигательной системы врачи выделяют несколько основных форм.
Все формы метафизарной дисплазии передаются двумя механизмами наследования:
- аутосомно-рецессивным;
- аутосомно-доминантным.
Это влияет на проявление, симптоматику и тип заболевания. Патология с одинаковой частотой встречается среди мужчин и женщин, в большинстве случаев проявляется в первые годы жизни. Некоторые типы болезни отличаются высокой степенью летальности.
Основные причины метафизарной дисплазии
У специалистов недостаточно информации о путях передачи заболевания. В большинстве случаев при изучении генетического материала и сборе анамнеза выявляется аутосомно-доминантная форма, которая связана с мутацией гена SFRP4 в 5 хромосоме. Он отвечает за кодирование белков, переносящих пирофосфат. Активное вещество необходимо для полноценных и активных процессов резорбции костной ткани. При его недостатке плотность кости уменьшается до критического уровня, меняются ее структура и форма.
Мутация белка-переносчика пирофосфата приводит к аномальному строению трубчатых длинных костей скелета. На генетическом уровне меняется процесс активности остеокластов.
Аутосомно-рецессивная форма болезни встречается реже, связана с аномалией генов в 7-й хромосоме. Мутация приводит к поражению белка, который принимает участие в формировании межклеточных контактов в костной ткани. Нарушается процесс передачи информации низкомолекулярными соединениями. Но врачам не удается до конца понять причину избирательного поражения метафиза костей.
Для аутосомно-рецессивной формы характерно поражение внутренних органов и тканей, в которых также присутствуют мутировавшие белки. У пациентов чаще наблюдаются вторичные нарушения, связанные с патологическим ростом костей и давлением на нервные окончания.
Метафизарная дисплазия у детей при аутосомно-рецессивной форме наследования проявляется, когда оба родителя являются носителями мутированного гена.

Виды заболевания
При метафизарной дисплазии происходит генетическое нарушение в строении костей скелета. При этом практически не меняется форма черепа, не возникают мутации нижней или верхней челюсти, характерные для краниометафизарной формы.
Заболевание сложно обнаружить без рентгенологического исследования. Среди характерных признаков – конусообразное расширение в трубчатых костях нижних конечностей. Они по форме начинают напоминать стеклянные колбы, используемые в лабораториях.
В некоторых медицинских источниках метафизарная дисплазия носит название "болезнь Пайла" – в честь ученого, который впервые описал патологию в 1931 году. На сегодняшний день существует 8 основных типов. Они различаются внешними симптомами и характером течения болезни:
- тип Янсена;
- Шмидта;
- МакКьюзика;
- метафизарная анадисплазия;
- дисплазия Швахмана-Даймонда;
- дисплазия с дефицитом аденозинд-дофаминазы по типу Шпар;
- метафизарная хондродисплазия (болезнь Пайла);
- метафизарная акроскифодисплазия.
Установить вид заболевания помогает комплексное обследование, включающее генетическое исследование биологического материала пациента.
Симптомы метафизарной дисплазии
Характерным признаком заболевания является сильное смещение трубчатых костей при одновременном и значительном расширении колбы Эрленмейера в ногах. Особенно симптомы выражены при поражении бедренной части. При метафизарной дисплазии негативные изменения в строении черепа минимальны. В редких случаях сильно выступают лобные доли над глазницами, создавая асимметричное строение лица.
При заболевании наблюдаются расширение грудной клетки, неправильное строение таза с искривлением, поражением тазобедренного сустава. Меняется угол наклона нижней челюсти, плечевые кости также по ширине превышают норму.
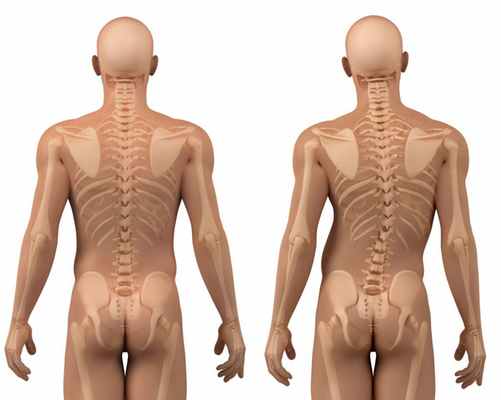
У многих больных сильная степень искривления позвоночника, тяжелые формы сколиоза. Даже в детском возрасте плотность костной ткани значительно меньше нормы, что приводит к остеопорозу. Кости становятся слишком хрупкими, с трудом выдерживают вес тела при ходьбе и активных движениях. Это основная причина частых патологических переломов ребер и нижних конечностей.
Не менее распространены симптомы метафизарной дисплазии:
- тяжелые формы кариеса с многочисленным поражением зубов в пришеечной части;
- аномальное строение околоносовых, лобных пазух;
- ограничение подвижности верхних конечностей в локтевом суставе;
- сильная мышечная слабость;
- гипотонус нижних конечностей;
- неправильный прикус;
- плоскостопие.
При некоторых формах заболевания быстро прогрессирует сколиоз. Происходит сдавливание нервных окончаний спинного мозга, которые иннервируют внутренние органы. Это одна из причин проблем с сердечно-сосудистой и эндокринной системами, органами пищеварения, головных и мышечных болей.
Метафизарная дисплазия типа Янсена
Крайне редкое заболевание: на сегодняшний день достоверно описано не более 10 случаев патологии. Среди характерных признаков, указывающих на этот тип болезни:
- широко расставленные глаза;
- развитие экзофтальма;
- неестественно широкая и укороченная переносица;
- недоразвитие нижней челюсти, которое приводит к неправильному прикусу, проблемам с зубами;
- низкий рост, связанный с укорочением бедренной кости.
При дисплазии Янсена происходит тяжелое поражение тазобедренных и коленных суставов. Постепенно нарастают сгибательные контрактуры в суставах нижних конечностей, что создает трудности в ходьбе, меняет походку, приводит к хромоте. Средний рост пациентов не превышает 120−125 см.
Искривление позвоночника прогрессирует по мере взросления, приводит к неестественному положению тела. Человек ходит, сильно наклонившись вперед, руки свисают, кисти могут доставать до колен. Суставы сильно увеличены в размере за счет патологического строения метафизов.
При метафизарной дисплазии Янсена показано хирургическое вмешательство. Необходимо восстановить оси конечностей для улучшения походки. Корректирующее вмешательство помогает повысить выносливость и улучшить качество жизни больного. Таким пациентам противопоказаны занятия спортом, связанные с переносом тяжестей, длительные пешие прогулки.
Метафизарная дисплазия типа Шмидта
При этом виде заболевании пациенты значительно отстают в росте. Наблюдается выраженная деформация нижних конечностей. Первые симптомы проявляются уже на 1-2-м году жизни. У детей появляется "утиная походка", сильная хромота. Они отказываются от активных игр, могут жаловаться на боли в коленных или тазобедренных суставах.
У пациента с типом Шмидта наблюдаются следующие патологии:
- выраженный поясничный лордоз;
- варусная деформация бедер;
- О-образная форма ног.
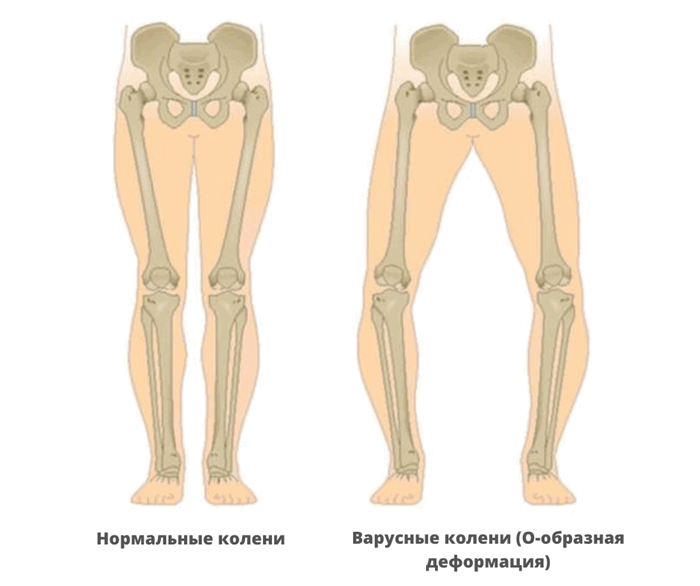
При диагностике врачи обращают внимание на характерное поражение проксимального отдела шейки бедра. Кости имеют неправильную форму, недостаточную плотность, низкое содержание кальция и микроэлементов. На рентгеновских снимках заметны неровные контуры, расширенные участки эпифизов.
При метафизарной дисплазии Шмидта не наблюдается поражение внутренних органов и систем. У больных практически отсутствуют аномалии в строении черепа и лица. Основное поражение приходится на нижние конечности и позвоночный столб.
По мере взросления проводится хирургическая коррекция. Она позволяет устранить выраженные деформации костей, сохранить здоровье суставов. При необходимости выполняется эндопротезирование.
Метафизарная дисплазия МакКьюзика
Метафизарная хондродисплазия Мак-Кьюсика (гипоплазия хряща и волос) – редкое заболевание, которое является причиной карликовости. У пациентов наблюдается сильное укорочение конечностей (до 20 см), что связано с дисплазией скелета.
Также для заболевания характерна предрасположенность к онкологическим опухолям, низкий уровень иммунитета. Основные признаки дисплазии МакКьюзика:
- короткие конечности;
- аномально низкий рост человека;
- тонкие светлые волосы, брови и ресницы;
- высокая степень растяжимости суставов рук и ног;
- заболевания позвоночника (сколиоз, лордоз, кифоз);
- дефектный иммунитет, связанный с недостатком антител.

Заболевание передается аутосомно-рецессивным путем. Пациенты часто страдают от клеточного иммунодефицита. Кроме карликовости и патологического строения скелета, у них наблюдается повышенная восприимчивость к вирусным, инфекционным заболеваниям, которые протекают с особой степенью тяжести. Дети с метафизарной дисплазией МакКьюзика часто погибают в раннем возрасте от обычной простуды или ветряной оспы.
Для пациентов с типом MакКьюзика характерны хроническая анемия, патологии кишечника. На рентгеновских снимках заметно увеличение метафизов в области коленного сустава. В детском возрасте нередко встречается двояковыпуклое строение позвонков, которое постепенно исчезает после взросления.
Акроскифодисплазия
У пациентов с этим типом заболевания отмечается сильная задержка роста. При этом метафизарные части трубчатых костей аномально большие, особенно в нижних конечностях. Одновременно нарушено строение диафиза в бедрах, развивается вальгусное искривление ног.
У больных наблюдается сильная косолапость, умеренная деформация трубчатых костей рук и фаланг кистей. Нередко заболевание приводит к брахидактилии, психомоторной задержке развития.
Некоторые дети появляются с аномально короткими ребрами, что приводит к врожденной асфиксии. Отмечаются укорочение основания черепа, аномалии развития гениталий, желудочно-кишечного тракта, анального отверстия, желудка. Некоторые патологические изменения несовместимы с жизнью.
Дисплазия Швахмана-Даймонда
При заболевании происходит поражение скелета человека, кроме того, возникают изменения в работе поджелудочной железы: снижается выработка ферментов, ответственных за расщепление жиров. Из-за патологического строения возникают дисфункция костного мозга, сколиоз и другие заболевания позвоночного столба.
Среди характерных симптомов:
- низкий рост;
- хроническая анемия;
- тяжелые патологии печени;
- гематологические нарушения;
- низкий уровень лейкоцитов.
Первые признаки проявляются на 5-6-м месяце жизни ребенка. У детей с синдромом Швахмана-Даймонда критически снижено количество тромбоцитов, эритроцитов. Они часто болеют простудными заболеваниями, бактериальными инфекциями, пневмониями, страдают хроническим средним отитом.
При недостаточности и липоматозе поджелудочной железы возникают проблемы с усвоением жиров, требующие постоянной медикаментозной терапии. При постоянном наблюдении врачей пациенты доживают до 18−20 лет.
Метафизарная дисплазия типа Спарк (Шпар)
Это одно из нескольких расстройств, которые раньше называли метафизарным дизостозом. Редкое заболевание, при котором у человека изогнуты ноги, возникает низкорослая карликовость. В некоторых случаях сгибание колен настолько сильно, что требует хирургической коррекции.
Тип Спарк по симптоматике и течению болезни во многом напоминает метафизарную дисплазию Шмидта. Патология имеет аутосомно-рецессивное наследование.
Болезнь Пайла
В отличие от других форм метафизарной дисплазии при болезни Пайла наблюдается аномальный рост трубчатых костей. У пациента появляется выраженная деформация коленных суставов, сколиоз 1-2-й степени. Уже в 8−9 лет у детей развиваются дисплазия тазобедренных суставов, поражение метафизов большеберцовых костей. Среди характерных симптомов:
- позднее выпадение молочных зубов, дисплазия эмали, увеличенные межзубные промежутки, склонность к кариесу;
- высокий рост;
- черный акантоз по линии роста волос;
- повышенная сухость кожного покрова;
- стрии в области сгибания суставов;
- конусовидные пальцы.
Такие дети значительно опережают в росте сверстников, часто сталкиваются с патологическими переломами в раннем возрасте. У многих пациентов наблюдаются патологии эндокринной системы, диффузный зоб. По мере роста больные жалуются на боли в суставах, которые ограничивают движения. Развиваются артроз и другие дегенеративные заболевания, влияющие на общее самочувствие.
Методы лечения
При некоторых формах метафизарной дисплазии у пациентов отмечается сильный дефицит костной массы. После определения плотности кости врач может порекомендовать курсы минерализующих препаратов на основе кальция, фтора, магния, витамина D. Они не меняют структуру, но снижают риск патологических переломов.
Любая медикаментозная терапия при метафизарной дисплазии носит вспомогательный характер, применяется для устранения симптомов. В современной медицине не существует препаратов, способных остановить патологическое изменение метафизарных частей костей. При болезни Пайла необходимо дополнительно отслеживать гормональный фон, корректировать его уровень.
При частых вывихах и подвывихах тазобедренного сустава рекомендуется ношение специальных протезов. Пациентам индивидуально изготавливают ортопедическую обувь для коррекции укороченной конечности. Для замедления развития сколиоза требуется корсет и постоянная лечебная физкультура. При отсутствии эффекта назначается операция.
Хирургическое лечение
Корригирующая остеотомия показана при деформации кости, помогает избавиться от дефекта и искривления кости. Она заключается в создании искусственного перелома, после которого отломки фиксируют в анатомически правильном положении.
Корригирующая остеотомия позволяет правильно распределить нагрузки на суставы и кости, снижает риск патологических переломов. Во время манипуляции хирурги могут убрать костные наросты, которые образовались при неправильном срастании, восстановить функциональность конечностей и позвоночника.
Операция проводится под общей анестезией. Хирург осторожно распиливает кость, после чего укрепляет и соединяет фрагменты с помощью наружной или внутренней фиксации. Если необходимо удлинить конечность, используется аппарат Илизарова.
Реабилитация занимает от 3 месяцев до года, позволяет избавиться от О и Х-образной деформации нижних конечностей, тяжелой степени кифоза, лордоза или сколиоза. Некоторым пациентам с тяжелой формой метафизарной дисплазии рекомендуется операция по устранению стеноза спинного мозга и суставных контрактур.
После операции для сохранения эффекта назначается комплексная реабилитация. Она может включать: лечебную физкультуру и массаж, физиотерапию для устранения мышечного напряжения или гипотонуса.
Прогноз для больного зависит от типа метафизарной дисплазии. Некоторые виды приводят к гибели плода во время беременности, имеют высокую степень летальности. Многие патологии при правильно подобранном лечении и проведении операции позволяют пациенту активно работать, вести обычный образ жизни.
Читайте также: