Гемохроматоз. Синдром Блума.
Добавил пользователь Alex Обновлено: 27.01.2026
Блума синдром — редкое генетическое заболевание, характеризующееся низкорослостью, повышенной чувствительностью кожи к ультрафиолетовым лучам от солнца. Синдром Блума является прототипом группы генетических состояний, известных как синдром поломок хромосом. Генетическая аномалия при синдроме Блума вызывает проблемы с ДНК, в результате чего большое количество хромосом перестраивается.
К внешним симптомам синдрома Блума относиться множественные мелкие сосуды на носу и щеках. Пациенты с данным синдромом страдают от частых инфекционных заболеваний из-за слабой иммунной системы. Тревожным отличием синдрома является заметно повышенная восприимчивость к развитию многих типов рака, особенно лейкемии, лимфомы и .
Синдром Блума наследуется как аутосомно-рецессивный генетический признак и входит в число генетических болезней ашкеназов.
Симптомы
Взрослые с синдромом Блума имеют недостаточный вес и маленький размер головы, но при этом пропорции тела в нормальном состоянии. Дети и новорожденные обычно появляются на свет с маленькой или узкой головой и лицом. Иногда эти признаки сопровождает красноватая сыпь лица, которые появляются из .
По неутешительной статистику, у 50% людей с этим расстройством в конечном итоге развивается один из множества злокачественных новообразований, чаще это лейкемия или рак желудка. У лиц с данным синдромом большая вероятность развития заболевания .
А иногда и является общим и главным признаком, люди с синдромом Блума не в состоянии производить сперму. Женское бесплодие также распространено, потому что менструация прекращается слишком рано.
Кроме того, люди с синдромом Блума, как правило, имеют нарушения иммунной системы, которые часто приводят к инфекционным заболеваниям, таким как пневмония, у пациентов часто наблюдается характерно высокий голос , не правильно растут зубы, лишний шестой палец или пальцы.
Причины
Синдром Блума наследуется как аутосомно-рецессивный генетический признак. Каузативный ген был сопоставлен хромосомном локусе 15q26.1 и отвечает за кодировку белка, известного как бислойные липидные мембраны. Одна мутация, отвечает почти за все случаи данного синдрома.
Генетические заболевания обусловлены сочетанием генов для одного определенного признака, которые находятся в хромосомах, полученных от отца и матери.
Рецессивные генетические нарушения происходят, когда человек наследует один и тот же ген для одного и того же признака от каждого родителя.
Сопутствующие заболевания при синдроме Блума
Симптомы следующих расстройств могут быть аналогичны симптомам синдрома Блума. Сравнения могут быть полезны для диагностики:
- Хромосомные синдромы нестабильности аутосомно-рецессивных наследственных заболеваний, связаны с повышенной хромосомной нестабильностью и генетической нестабильностью, ставят пораженных лиц на более высокий уровень вероятности к заболеванию: анемия Фанкони, атаксия-телеангиэктазия, и пигментная ксеродерма.
- Синдром Коккейна — представляет собой переменную генетическое заболевание, характеризующееся дефицитом роста и прогрессивным неврологическое ухудшением.
- Синдром Ротмунда-Томсона — это генетическая, полисистемная патология, которая, как правило, обнаруживается на ранних этапах, после рождения ребенка. Во время раннего младенчества у людей с синдромом Ротмунда-Томсона развиваются красные, воспаленные бляшки на коже, сопровождающиеся скоплениями жидкости между слоями ткани под кожей, проще говоря отеком.
Лечение
Лечение синдрома поддерживающее, поскольку не существует узконаправленной терапии на лечение синдрома Блума. Солнцезащитные средства могут быть использованы для снижения воздействия ультрафиолетового излучения, а также всем лицам с данным синдромом следует ограничить контакт с прямыми солнечными лучами. Также рекомендуются периодические походы к
Синдром Блума – редкое наследственное, аутосомно-рецессивное заболевание, характеризующееся телеангиэктатической эритемой лица, повышенной фоточувствительностью, задержкой роста.
Причины синдрома Блума
Заболевание обусловлено аутосомно-рецессивным геном. Возникает в семьях с высокой частотой инбридинга. Чаще поражаются лица мужского пола.
Симптомы синдрома Блума
Главными признаками заболевания являются эритема лица и карликовый рост. Эритема развивается в грудном или раннем детском возрасте в виде пятен или бляшек, весьма напоминающих красную волчанку. Чаще они группируются в виде бабочки на носу и щеках, но могут появляться на веках, лбу и ушных раковинах, а иногда на тыле кистей и предплечий. Поверхность их слегка шелушится.
Обычно после воздействия солнечных лучей наблюдается экзацербация высыпаний, а также появление пузырей, кровотечения и корочек на губах.
Диагностика синдрома Блума
Диагноз основывается на клинической картине, а также выявлении патоло¬гии репарации ДНК, высокой частоты сестринских хроматидных обменов, изменений лимфоцитов. При пренатальной диагностике используют оценку сестринских хроматидных обменов.
Лечение синдрома Блума
Лечение симптоматическое: фотозащитные средтства, каротиноиды, витамин Е, иммунокорректоры. В качестве профилактики заболевания рекомендуется избегать кровнородственных браков.
Синдром Блума Bloom syndrome - синдром Блума.
НЗЧ, характеризующееся недоразвитием (карликовостью), ослабленным иммунитетом, повышенной чувствительностью к солнечному свету, дистрофическими изменениями кожи, в ядрах клеток может происходить пульверизация хроматина; в основе С.Б. - множественная ломкость хромосом ; наследуется по аутосомно-рецессивному типу.
(Источник: «Англо-русский толковый словарь генетических терминов». Арефьев В.А., Лисовенко Л.А., Москва: Изд-во ВНИРО, 1995 г.)
Смотреть что такое "синдром Блума" в других словарях:
синдром Блума - НЗЧ, характеризующееся недоразвитием (карликовостью), ослабленным иммунитетом, повышенной чувствительностью к солнечному свету, дистрофическими изменениями кожи, в ядрах клеток может происходить пульверизация хроматина; в основе С.Б.… …
Блума синдром - * Блума сіндром * Bloom syndrome наследственное заболевание аутосомно рецессивного типа, проявляющееся в виде телеангиэктатической эритемы на лице в виде бабочки, иногда на руках и предплечиях. Пациенты карлики с сохраненной нормальной… … Генетика. Энциклопедический словарь
- (D. Bloom, род. в 1892 г., амер. дерматолог) сочетание недоразвития скелета, гипофизарной карликовости и гипогонадизма с врожденной телеангиэктатической эритемой лица, участками кератоза и гиперпигментации на туловище; проявляется в детском… … Большой медицинский словарь
ДЕФЕКТЫ РЕПАРАЦИИ ДНК - мед. Репарация клеточный механизм коррекции повреждённой последовательности ДНК (точечные мутации, делеции, структурные нарушения и др.). Системы репарации Темновая (фотореактивация): удаляются УФ индуцированные ковалентные связи между смежными … Справочник по болезням
Теломераза фермент, добавляющий особые повторяющиеся последовательности ДНК (TTAGGG у позвоночных) к 3 концу цепи ДНК на участках теломер, которые располагаются на концах хромосом в эукариотических клетках. Теломеры содержат уплотненную ДНК … Википедия
Chromosome fragility ломкость хромосом. Форма хромосомных аномалий, проявляющихся в виде гэпов , которые, как правило, локализованы в определенных участках хромосом ломких сайтах (обозначаются fra); признак Л.х. в определенном сайте… … Молекулярная биология и генетика. Толковый словарь.
ломкость хромосом - Форма хромосомных аномалий, проявляющихся в виде гэпов, которые, как правило, локализованы в определенных участках хромосом ломких сайтах (обозначаются fra); признак Л.х. в определенном сайте передается согласно Менделевским законам; Л.х.… … Справочник технического переводчика
Bloom syndrome. См. синдром Блума. (
Гемохроматоз (син.: бронзовый диабет, пигментный цирроз печени, синдром Труазье- Ано-Шоффара) - аутосомно-рецессивное наследственное заболевание, характеризующееся накоплением в организме железа. Ген гемохроматоза расположен в 6-й хромосоме и сцеплен с локусом системы HLA. Главное звено патогенеза - усиление всасывания железа в тонкой кишке и накопление его, в первую очередь, в печени, поджелудочной железе, сердце, половых железах и коже. Гемохроматоз является сравнительно частой причиной цирроза печени, сахарного диабета, гипогонадизма, артрита, кардиомиопатии.
Наиболее важный клинический симптом гемохроматоза - поражение кожи в виде серо-коричневой пигментации, которая усиливается под воздействием солнечного облучения. Однако степень ее выраженности не коррелирует с нередко выявляемым отложением железа вокруг эккринных потовых желез и накоплением меланина в базальном слое эпидермиса. Кожными проявлениями гемохроматоза также могут быть сухость кожи, мелкопластинчатое шелушение, ихтиоз, звездчатые телеангиэктазии, эритроз ладоней, алопеция, койлонихия, очаговая атрофия кожи. Гепатоцеллюлярный рак развивается как позднее осложнение в 7-37% случаев и является основной причиной смерти. Обычно он возникает у пожилых людей с циррозом печени.
Гистологически при гемохроматозе выявляют значительное отложение меланина в базальном слое эпидермиса. В дерме, преимущественно вокруг придатков кожи, определяется скопление больших масс гемосидерина.
Гемохроматоз можно заподозрить при повышении уровня сывороточного ферритина и трансферрина. Уровень же сывороточного железа подвержен значительным колебаниям, в связи с чем не является диагностически ценным. Диагноз подтверждают путем биопсии печени с определением отложений железа в клетках печени. Заподозрить гепатоцеллюлярный рак позволяют ультразвуковое исследование, компьютерная томография, определение уровня сывороточного а-ферритина.
Наиболее эффективно кровопускание при гемохроматозе . Проводится лечение цирроза печени, сахарного диабета и другой выявленной патологии. Рекомендуется медико-генетическое консультирование.
Синдром Блума
Синдром Блума (син.: врожденная телеангиэктатическая эритема; врожденная телеангиэктатическая эритема и нанизм) - редкое наследственное, аутосомно-рецессивное заболевание, характеризующееся телеангиэктатической эритемой лица, повышенной фоточувствительностью, задержкой роста. Возникает в семьях с высокой частотой инбридинга. Чаще поражаются лица мужского пола. Культура лимфоцитов и фибробластов больных отличается высокой частотой хромосомных аберраций и повышенной чувствительностью к УФ-облучению, отмечена недостаточность фермента ДНК-лигазы I. Клетки при синдроме Блума проявляют хромосомную нестабильность, приводящую к высокому уровню соматических мутаций как внутриутробно, так и в постнатальной жизни. У больных с синдромом Блума отмечаются нарушения со стороны В-(снижение уровня IgA и IgM) и Т-системы иммунитета. В частности, повышение онкологической заболеваемости при синдроме Блума связывают с подавлением пролиферативной реакции лимфоцитов на митогены. В то же время связь дефекта иммунной функции с недостаточностью энзима, участвующего в репарации ДНК-лигазы I, остается неясной. Ген синдрома Блума пока не клонирован. Соотношение частоты различных типов рака при синдроме Блума, отражающее их соотношение в обшей популяции, подтверждает, что синдром Блума является мутирующим фенотипом, при котором каждая клетка тела более склонна к соматической мутации. При этом сопутствующий иммунодефицит неясного происхождения может также играть роль в опухолевой прогрессии.
Синдром Блума начинается в первые недели жизни с появления эритемы, пузырей и корок на щеках, ушных раковинах, спинке носа, позже - на тыле кистей. Обычно отмечается кожная фоточувствительность даже после минимального ультрафиолетового облучения, которая нередко сопровождается развитием телеангиэктазий и повышенной ранимостью кожи. Позже на облучаемых и необлучаемых участках кожи появляются очаги атрофии, крапчатой гиперпигментации и гипопигментации, а также пятна типа «кофе с молоком». Обострение происходят, как правило, весной и летом. Дети обычно рождаются с низкой массой тела и в дальнейшем отстают в физическом развитии, хотя умственное развитие не страдает. Отмечаются диспропорционально узкий (доли-хоцефалический) череп, выступающий нос и маленькая нижняя челюсть (птичье лицо). Встречаются и другие аномалии развития: клиндактилия, сандактилия, полидактилия, деформация стоп, врожденные пороки сердца, дефекты зубов. Многие больные низкорослы, имеют высокий голос. Продолжительность жизни короткая.
Гистологическая картина синдрома Блума неспецифична: уплощение и атрофия эпидермиса, расширение сосудов сосочкового слоя дермы, периваскулярный лимфоцитарно-гистиоцитарный инфильтрат.
Диагноз синдрома Блума устанавливается на основании клинической картины, выявления патологии репарации ДНК, высокой частоты сестринских хроматидных обменов, а также изменений в лимфоцитах. В качестве метода пренатальной диагностики заболевания рекомендуется использовать оценку сестринских хроматидных обменов.
Дифференциальный диагноз синдрома Блума проводится с красной волчанкой, синдромом Коккейна, порфирией, синдромом Ротмунда-Томсона.
При лечении синдрома Блума применяются фотозащитные средства, каротиноиды, витамин Е, иммунокорректоры. Больные должны наблюдаться дерматоонкологом с целью ранней диагностики немеланотических раков кожи. В качестве профилактики заболевания следует избегать близкородственных браков.
Гемохроматоз. Синдром Блума.
Гемохроматоз. Синдром Блума.
Гемохроматоз (син.: бронзовый диабет, пигментный цирроз печени, синдром Труазье— Ано—Шоффара) — аутосомно-рецессивное наследственное заболевание, характеризующееся накоплением в организме железа. Ген гемохроматоза расположен в 6-й хромосоме и сцеплен с локусом системы HLA. Главное звено патогенеза — усиление всасывания железа в тонкой кишке и накопление его, в первую очередь, в печени, поджелудочной железе, сердце, половых железах и коже. Гемохроматоз является сравнительно частой причиной цирроза печени, сахарного диабета, гипогонадизма, артрита, кардиомиопатии.
Наиболее важный клинический симптом гемохроматоза — поражение кожи в виде серо-коричневой пигментации, которая усиливается под воздействием солнечного облучения. Однако степень ее выраженности не коррелирует с нередко выявляемым отложением железа вокруг эккринных потовых желез и накоплением меланина в базальном слое эпидермиса. Кожными проявлениями гемохроматоза также могут быть сухость кожи, мелкопластинчатое шелушение, ихтиоз, звездчатые телеангиэктазии, эритроз ладоней, алопеция, койлонихия, очаговая атрофия кожи. Гепатоцеллюлярный рак развивается как позднее осложнение в 7-37% случаев и является основной причиной смерти. Обычно он возникает у пожилых людей с циррозом печени.
Гистологически при гемохроматозе выявляют значительное отложение меланина в базальном слое эпидермиса. В дерме, преимущественно вокруг придатков кожи, определяется скопление больших масс гемосидерина.
Гемохроматоз можно заподозрить при повышении уровня сывороточного ферритина и трансферрина. Уровень же сывороточного железа подвержен значительным колебаниям, в связи с чем не является диагностически ценным. Диагноз подтверждают путем биопсии печени с определением отложений железа в клетках печени. Заподозрить гепатоцеллюлярный рак позволяют ультразвуковое исследование, компьютерная томография, определение уровня сывороточного а-ферритина.
Наиболее эффективно кровопускание при гемохроматозе. Проводится лечение цирроза печени, сахарного диабета и другой выявленной патологии. Рекомендуется медико-генетическое консультирование.

Синдром Блума
Синдром Блума (син.: врожденная телеангиэктатическая эритема; врожденная телеангиэктатическая эритема и нанизм) — редкое наследственное, аутосомно-рецессивное заболевание, характеризующееся телеангиэктатической эритемой лица, повышенной фоточувствительностью, задержкой роста. Возникает в семьях с высокой частотой инбридинга. Чаще поражаются лица мужского пола. Культура лимфоцитов и фибробластов больных отличается высокой частотой хромосомных аберраций и повышенной чувствительностью к УФ-облучению, отмечена недостаточность фермента ДНК-лигазы I. Клетки при синдроме Блума проявляют хромосомную нестабильность, приводящую к высокому уровню соматических мутаций как внутриутробно, так и в постнатальной жизни. У больных с синдромом Блума отмечаются нарушения со стороны В-(снижение уровня IgA и IgM) и Т-системы иммунитета. В частности, повышение онкологической заболеваемости при синдроме Блума связывают с подавлением пролиферативной реакции лимфоцитов на митогены. В то же время связь дефекта иммунной функции с недостаточностью энзима, участвующего в репарации ДНК-лигазы I, остается неясной. Ген синдрома Блума пока не клонирован. Соотношение частоты различных типов рака при синдроме Блума, отражающее их соотношение в обшей популяции, подтверждает, что синдром Блума является мутирующим фенотипом, при котором каждая клетка тела более склонна к соматической мутации. При этом сопутствующий иммунодефицит неясного происхождения может также играть роль в опухолевой прогрессии.
Синдром Блума начинается в первые недели жизни с появления эритемы, пузырей и корок на щеках, ушных раковинах, спинке носа, позже — на тыле кистей. Обычно отмечается кожная фоточувствительность даже после минимального ультрафиолетового облучения, которая нередко сопровождается развитием телеангиэктазий и повышенной ранимостью кожи. Позже на облучаемых и необлучаемых участках кожи появляются очаги атрофии, крапчатой гиперпигментации и гипопигментации, а также пятна типа «кофе с молоком». Обострение происходят, как правило, весной и летом. Дети обычно рождаются с низкой массой тела и в дальнейшем отстают в физическом развитии, хотя умственное развитие не страдает. Отмечаются диспропорционально узкий (доли-хоцефалический) череп, выступающий нос и маленькая нижняя челюсть (птичье лицо). Встречаются и другие аномалии развития: клиндактилия, сандактилия, полидактилия, деформация стоп, врожденные пороки сердца, дефекты зубов. Многие больные низкорослы, имеют высокий голос. Продолжительность жизни короткая.
Гистологическая картина синдрома Блума неспецифична: уплощение и атрофия эпидермиса, расширение сосудов сосочкового слоя дермы, периваскулярный лимфоцитарно-гистиоцитарный инфильтрат.
Диагноз синдрома Блума устанавливается на основании клинической картины, выявления патологии репарации ДНК, высокой частоты сестринских хроматидных обменов, а также изменений в лимфоцитах. В качестве метода пренатальной диагностики заболевания рекомендуется использовать оценку сестринских хроматидных обменов.
Дифференциальный диагноз синдрома Блума проводится с красной волчанкой, синдромом Коккейна, порфирией, синдромом Ротмунда—Томсона.
При лечении синдрома Блума применяются фотозащитные средства, каротиноиды, витамин Е, иммунокорректоры. Больные должны наблюдаться дерматоонкологом с целью ранней диагностики немеланотических раков кожи. В качестве профилактики заболевания следует избегать близкородственных браков.
Гемохроматоз. Синдром Блума.

Гемохроматоз (синдром железного человека)
Принято считать, что дефицит железа в организме есть чуть ли не у каждой женщины и у каждого второго мужчины. Такие выводы мы делаем, когда слышим заявления о том, что нужно побольше употреблять в пищу продуктов, богатых этим элементом. На самом деле переизбыток железа не менее опасен, чем его дефицит.
О гемохроматозе рассказывает Давид Юрьевич Матевосов, заведующий отделением персонифицированной медицины, руководитель МЕДСИ Premium, врач-гастроэнтеролог, гепатолог, к. м. н.
«Гемохроматоз – это патологические состояния, связанные с разными причинами, при которых организм усваивает слишком много железа из пищи. Избыток железа может накапливаться в органах, приводя к нарушению их работы и возникновению заболеваний. Особенно этому подвержены печень, сердце и поджелудочная железа. Слишком много железа может привести к опасным для жизни состояниям, таким как цирроз печени, сердечная недостаточность и сахарный диабет.
Принято разделять гемохроматоз на первичный, т. е. наследственный, и вторичный, приобретенный. В первом случае речь идет о дефектах в генах, отвечающих за работу ферментных систем, регулирующих транспорт железа. В этой ситуации белок – переносчик железа, а именно ферритин способствует увеличению запасов железа в тканях. Виной тому – дефекты в генах, которые могут передаваться по наследству. Такие генетические особенности встречаются у 10% населения в целом, но далеко не все эти люди в итоге приходят к развитию болезней. Ведь гены могут быть изменены частично (в подавляющем большинстве случаев), а могут быть полностью дефектны (всего 0,33%), когда риск возникновения клинически выраженного заболевания гораздо выше.
Вторичный гемохроматоз, скорее даже синдром перегрузки железом, является следствием других болезней, например, длительного переливания крови при хронических и редких анемиях, алкогольной болезни печени, кожной порфирии. В этом случае организм другими механизмами усваивает избыток поступающего железа или создаются условия, при которых клетки крови вырабатываются с избыточным содержанием гема (т. е. железа).
«Железного человека» можно узнать по специфической окраске кожи. Это не цвет загара, а коричнево-бронзовый оттенок. Если генетические дефекты уже привели к развитию заболеваний, в частности печени, врач может увидеть так называемые печеночные знаки – сосудистые звездочки на коже носа, щек, шеи, груди и красные «печеночные» ладони, иногда с четкой очерченностью.
В организм человека железо поступает в 2 вариантах – двухвалентном и трехвалентном. Двухвалентное железо (гемовое или закисное) содержится только в пище животного происхождения (красное мясо, печень, моллюски) и усваивается в тонкой кишке практически полностью. Большую часть дневного рациона составляет негемовое (окисное) или трехвалентное железо, которое содержится преимущественно в пище растительного происхождения. Около 60% всего железа находится в гемоглобине, а примерно 30% железа входит в состав ферритина – внутренних запасов железа. На самом деле доля поступающего с пищей железа – это лишь малая часть железа, которое содержится в организме. Наш организм способен обратно всасывать железо из отживших свой срок красных телец крови – эритроцитов, за счет чего поддерживается практически постоянный объем нужного организму железа. То есть организм способен повторно использовать то железо, которое уже поработало в виде гемоглобина на эритроцитах. У женщин расход и потеря железа больше, чем у мужчин, поэтому и требуется его извне больше.
При гемохроматозе нет необходимости исключать полностью продукты – источники гемового и негемового железа ввиду описанных механизмов его циркуляции в организме. Но переусердствовать тоже нельзя. Как всегда, хорош принцип золотой середины.
Добавки, содержащие железо, теоретически могут способствовать большему всасыванию железа, особенно при бесконтрольном их применении. Известен исторический факт: употребление пива, приготовленного в стальных барабанах, явилось причиной синдрома перегрузки железом. Эффективность БАДов весьма сомнительна, в том числе и для коррекции железодефицитных состояний. Например, развеяны мифы о том, что совместное применение витамина C и препаратов железа у пациентов с железодефицитной анемией усиливает всасывание последнего. По факту витамин не оказывал влияния на всасывание железа и последующий уровень гемоглобина. Любителям поливитаминов необходимо знать, что кальций и железо, попадая в организм одновременно, конкурируют за усвоение. Железо усваивается на 45% лучше, если принимать его отдельно от кальция. Может увеличивать всасывание железа совместный прием его с витамином А.
Симптомы заболевания зависят от органа, который более всего поражен, однако практически все пациенты жалуются на значительную слабость и быструю утомляемость. Никаких специфических симптомов гемохроматоза не существует. Чаще всего диагноз ставится на стадии заболевания, когда уже пострадали несколько систем. От первых симптомов заболевания до постановки диагноза проходит обычно не менее десяти лет. Приблизительно у 50% пациентов с симптомами наследственного гемохроматоза наблюдается бронзовый сахарный диабет. Поражение суставов при гемохроматозе проявляется в виде болей мелких суставов кистей (обычно указательного и среднего пальцев). Отложение железа в волокнах сердечной мышцы и клетках проводящей системы сердца может привести к нарушению ритма сердца и развитию дилятационной кардиомиопатии с развитием в дальнейшем сердечной недостаточности. При гемохроматозе возможно развитие гипогонадизма и, соответственно, импотенции.
Печень также страдает не реже, в результате накопления железа постепенно формируется цирроз. Кроме того, случаи первичного рака печени без выявленных диффузных болезней печени относятся к неблагоприятным исходам наследственного гемохроматоза.
Чаще всего наследственный или вторичный гемохроматоз (особенно при алкогольной болезни печени) диагностируется именно врачом-гепатологом. Поиск возможных причин заболевания печени приводит к подтверждению высокого уровня ферритина сыворотки крови, повышения насыщения трансферрина железом. Эти изменения являются поводом для назначения врачом генетического теста на выявление наследственного гемохроматоза.
В ходе обследования и поиска причин патологии печени нередко наследственный гемохроматоз выявляется как фоновое состояние, которое может снижать эффективность лечения от другого заболевания печени: например, в век интерферонотерапии хронического гепатита С наследственный гемохроматоз негативно влиял на эффективность терапии и ухудшал прогнозы основного заболевания.
Изучение обмена железа как для женщин, так и для мужчин входит в перечень обычных скрининговых лабораторных тестов. Кроме того, содержание гемоглобина в периферической крови, размер эритроцитов могут дать нам косвенную информацию о состоянии обмена железа и повод для дальнейшего его детального изучения.
Настоятельно рекомендую не проводить диагностику по интернету, а обратиться к специалисту. И не назначать себе БАДы и препараты, не разобравшись в деталях!»
Мы не устаём повторять, что при выявлении у себя тех или иных симптомов необходимо проконсультироваться с врачом. Каждый из нас уникален, обладает своим набором генетических характеристик и перенесённых заболеваний. Если у вас есть сомнения в вопросах здоровья, обратитесь за помощью к специалистам.
Генетические заболевания : Аутосомно-рецессивные болезни
Синдром Блума (СБ), (англ. Bloom syndrome), также известен как синдром Блума-Торре-Махачека - это редкое аутосомно-рецессивное хромосомное расстройство, которое характеризуется высокой частотой разрывов и перестроек в хромосомах больного человека. Этот синдром был впервые описан врачом дерматологом Девидом Блумом (Dr. David Bloom) в 1954 году.
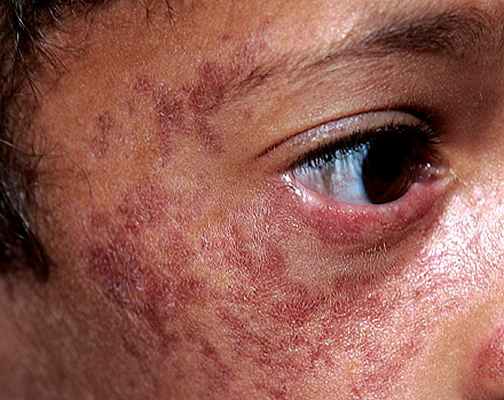
Лица, больные ВБ, как правило не высокого роста, с характерными высыпаниями на коже, которыевозникают почти сразу после первого воздействия солнечных лучей. Эти высыпания, как правило возникают на щеках (вызывая покраснение) и имеют форму бабочки. Но сыпь можетт возникать не только на лице, но и на других частях тела, на которые попадали солнечные лучи (спина, руки и т.д.).
Другие клинические признаки:
- высокий голос;
- специфические черты лица, такие как - длинно е и узкое лицо, микрогнатия нижней челюсти, большой нос и уши, изменения пигментации кожи, включая появление гипо-и гипер-пигментных пятен и родинок;
- телеангиэктазия (расширенные кровеносные сосуды), которые могут проявляться на коже, преимущественно на верхней части груди и спины;
- умеренным иммунодефицитом, который характеризуется дефицитом некоторых классов иммуноглобулинов, который, как считают исследователи, приводит к рецидивам пневмонии и инфекций ушей;
- гипогонадизм - это патологическое состояние, обусловленное недостаточной секрецией андрогенов, то есть организм мужчин, не может производить сперму, а значит, пораженные этим заболеванием лица мужского пола - бесплодны. У женщин эта болезнь проявляется преждевременным прекращением менструаций (преждевременная менопауза), т.е. они имеют пониженную способность к рождению детей. Однако, некоторые женщины, которые болеют синдром Блума могут иметь детей.
Осложнения, возникающие в результате воздействия заболевания, могут включать в себя хронические болезни легких, сахарный диабет, возникновение трудностей при обучении. У некоторых лиц, возникает умственная отсталость. Наиболее опасным осложнением можно назвать повышение вероятности возникновения опухолевых заболеваний.
Связь с возникновением рака
Вследствие большого количества мутаций, происходящих при заболевании синдромом Блума, у больныхсущественно повышается риск заболеть раком.
Предрасположенность к онкологическим заболеваниям характеризуется:
1) широким спектром проявления болезней, включающий лейкозы, лимфомы и карциномы;
2) ранним возрастом возникновения опухолевых заболеваний, поровну к остальному населению;
У лиц с синдромом Блума рак может возникнуть в любом возрасте. Средний возраст больных - 25 лет.
Патофизиология
Мутации в гене BLM, который является составной ДНК геликаз (хеликазы), вызывают возникновение синдрома Блума. ДНК геликазы (хеликазы) - это ферменты, которые раскручивают двухцепную спираль ДНК. Процесс «раскрутки» состоит из большого количества процессов, таких как синтез копий ДНК, транскрипция РНК, репарация ДНК и т.д.
Когда клетка готовится к делению, то хромосомы дублируются таким образом, что каждая новообразованная структура получает полный набор хромосом. Процесс удвоения называется - репликацией ДНК. Ошибки, допущенные при репликации ДНК могут привести к мутации. Белок BLM играет важную роль в поддержании стабильности ДНК в процессе репликации. Мутации гена BLM, связанные с синдромом Блума дезактивируют белок, который кодируется геном BLM и входит в состав ДНК хеликазы, или же нарушают процесс экспрессии (образование белка). Другими словами не происходит экспрессия этого гена. Отсутствие BLM белка или низкая его активность приводит к увеличению количества мутаций, именно поэтому молекулярные механизмы, с помощью которых BLM поддерживает стабильность хромосом, до сих пор остаются предметом для многих исследований.
У лиц, с СБ имеет место большое количество обменов между гомологичными хромосомами или сестринскими хроматидами (две молекулы ДНК, образующиеся в процессе репликации ДНК), в результате которых существенно увеличивается количество мутаций. Кроме того, на много чаще (в отношении лиц, не имеющих синдрома Блума) случается повреждение хромосом и их перестройка.
Сегодня ученые работают над тем, что установить (или опровергнуть) наличие прямой связи между молекулярными процессами, в которых участвует ген BLM и изменением хромосом. Кроме того, связь между молекулярными дефектами в клетках лиц с СБ, хромосомными мутациями, которые накапливаются в соматических клетках (всех клетках тела, кроме половых) и многими клиническими признаками синдрома Блума также является одним из направлений клинических исследований.
Синдром Блума наследуется по аутосомно-рецессивному типу. То есть, оба родителя должны быть носителями для того, чтобы их ребенок заболел. Носителями заболевания среди евреев Ашкенази, преимущественно являются выходцами из Восточной Европы, является 1 человек на 100. Если оба родителя являются носителями СБ, то в 25% случаев, ребенок может быть больным.
Тем членам семей, которые могут быть носителями заболевания рекомендуется обратиться с просьбой о генетической консультации или пройти генетическое тестирование. Для носителей-родителей, которые планируют рожать ребенка следует провести пренатальную диагностику с помощью цитогенетических и молекулярных методов. Также возможно осуществить молекулярный анализ ДНК для определения наличия мутаций.
Другие мутации при синдроме Блума
Впервые две миссенс мутации фермента пируват киназы М2 (H391Y и K422R), обнаружили в клетках больных синдромом Блума, как потом подтвердилось они повышают предрасположенность к возникновению рака. Результаты показывают, что несмотря на наличие мутаций в области домена мижсубединичного контакта, мутантные белки K422R и H391Y сохраняют свою гомотетраметрическую структуру, что делает их похожими по структуре на обычные белки, но их активность существенно уменьшается на 75 и 20% соответственно. Интересно, что H391Y показал 6-кратное увеличение родства со своим субстратом фосфоэнолпируватом и вел себя как неалостерический белок с нарушенным соединением (с субстратом).
Однако, K422R теряет свое родство с фосфоэнолпируват. В отличие от K422R, H391Y проявляет улучшенные тепловые характеристики и стабильность в определенных диапазонах значений рН, кроме того наблюдается меньшее влияние алостеричних ингибиторов Phe, и устойчивость во время структурных изменений при связывании активатора (фруктозо 1,6-бисфосфата) и ингибитора (Phe ). Оба мутанты показали небольшой сдвиг в оптимуме рН от 7,4 до 7,0.Совместная экспрессия гомотетрамеров дикого типа и мутанта PKM2 в клеточной среде, которая возникает в результате взаимодействия между ними на уровне мономеров, была позднее подтверждена в ходе клинических экспериментов. Кросс-мономерное взаимодействие существенно изменяет олигомерное состояние PKM2, через усиление димеризации и гетеротетрамеризации Эти особенности, как предполагается, играют важную роль при опухолевой прогрессии. PKM2 (M2-PK); потенциал многофункционального белка. Высказано предположение, что аберрантный белок PKM2 может быть основной причиной возникновения других симптомов, характерных для синдрома Блума.
"Статус носителя наследственных заболеваний"

Анализ "Статус носителя наследственных заболеваний" позволяет выявить гены 76 тяжелых наследственных заболеваний при планировании беременности или на ранних ее сроках, что позволяет предотвратить рождение ребенка с тяжелыми патологиями ( например: гемофилия, муковисцидоз, фенилкетонурия и др.). Спектр заболеваний приведен в таблице:
Боковой амиотрофический склероз
Дефицит 3-гидрокси-3-метилглутарил- КоА-лиазы
Полигландулярная аутоиммунная болезнь
Синдром Барттера 4А типа
Недостаточность среднецепочечной ацил- КоА-дегидрогеназы жирных кислот
Муколипидоз II типа
Муколипидоз III типа
Муколипидоз IV типа
Множественный дефицит карбоксилазы
Синдром Дабина — Джонсона
Синдром Элерса — Данлоса, дерматоспараксис
Врожденный дефицит протромбина
Недостаточность фактора XI
Rh null синдром
Синдром слабости синусового узла
Гликогенозы (болезнь накопления гликогена)
Несиндромальная нейросенсорная тугоухость
Тирозинемия I типа
Несиндромальная нейросенсорная тугоухость (DFNB59)
Недостаточность длинноцепочечной ацил-КоА-дегидрогеназы
Статьи для пациентов
Кто такой эмбриолог и чем он занимается? Для начала проведения процедуры оплодотворения in vitro, эмбриолог собирает семенную жидкость и проводит анализ эякулята.
ЭКО и здоровье ребенка: развенчиваем мифы Один из наиболее распространенных мифов – ЭКО оказывает негативные последствия на развитие и здоровье ребенка.
Внутриматочная инсеминация: показания к проведению и особенности лечения Искусственная инсеминация (ВМИ) – безболезненная технология лечения бесплодия путем введения предварительно обработанной в лаборатории спермы партнера или донора в канал шейки матки или напрямую в матку.
ЭКО и онкология: как забеременеть после химиолучевой терапии и других методов лечения Можно ли забеременеть, если у пары диагностирована онкология? Репродуктологи углубились в суть вопроса и нашли ответ: да, беременность возможна.
Орхиэктомия: показания, виды операции, последствия Орхиэктомия – оперативный метод лечения, предполагающий удаление одного или обеих яичек, если другие способы нерезультативны.
Способы диагностики и методы лечения аденомиоза матки Не всегда эндометриоз, как одну из причин бесплодия, можно выявить быстро и легко. Для диагностики применяются современные методики: УЗИ, МРТ, КТ органов малого таза и трансвагинальный датчик. Наиболее точное диагностирование достигается с помощью лапароскопии.
Аденомиоз и бесплодие: причины, симптомы и взаимосвязь Одним из наиболее распространенных и одновременно загадочных заболеваний в гинекологии является эндометриоз. Ему подвержено около 10% женщин репродуктивного возраста 25-35 лет, из чего вытекает повышенная актуальность проблемы.
Влияние новой вирусной инфекции на сперматозоиды Вспышка новой вирусной инфекции, согласно клиническим данным, приводит не только к респираторным заболеваниям и поражает лёгкие, но также вызывает гистопатологические и функциональные нарушения в различных органах, таких как яички, а также мужские половые пути.
Почему не получается забеременеть от ЭКО? Как показывает статистика, этот показатель является вполне закономерным, ведь и в естественном цикле не каждая женщина способна забеременеть сразу же после первой близости.
Читайте также: