Генетика гомоцистинурии. Наследование
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Энзим цистатион-β-синтаза участвует в преобразовании метионина в цистеин.
1) а, б, в;
2) а, в;+
3) а, г;
4) б, в.
2. Возможный побочный эффект пиридоксина у детей:
1) брадикардия;
2) дыхательные расстройства;+
3) конъюнктивит;
4) тахикардия.
3. Выберите тип наследования у больного с гомоцистинурией:
1) Х-сцепленное рецессивное;
2) аутосомно-доминантный;
3) аутосомно-рецессивный;+
4) сцепленное с Х-хромосомой доминантное наследование.
4. Выделяют следующие клинико-генетические формы классической гомоцистинурии:
1) В6-зависимая и В6-резистентная гомоцистинурия;+
2) гомоцистинурия, обусловленная нарушением активности 5-метилтетрагидрофолат-гомоцистеин-метилтрансферазы;+
3) гомоцистинурия, обусловленная нарушением активности N(5,10)-метилентетрагидрофолатредуктазы;+
4) отсутствует.
5. Выполнение офтальмоскопии при подозрении на эктопию хрусталика, соответствует силе рекомендаций:
1) D;
2) А;
3) В;+
4) С.
6. Ген, кодирующий синтез цистатион–β-синтазу локализован на длинном плече:
1) 10-й хромосомы;
2) 11-й хромосомы;
3) 15-й хромосомы;
4) 21-й хромосомы.+
7. Гомоцистин активирует:
1) протромбин (фактор II);
2) фактор Стюарта-Прауэра (фактор Х);
3) фактор Флетчера (ХIV фактор);
4) фактор Хагемана (ХII фактор).+
8. Гомоцистин активируя фактор Хагемана, тем самым:
1) может оседать в патологически измененной интиме сосуда;+
2) обладает высокой растворимостью;
3) обладает низкой растворимостью;+
4) способствует процессу тромбообразования.+
9. Гомоцистинурию от синдрома Марфана отличают следующие признаки:
1) Х-сцепленный рецессивный тип наследования;
2) более тяжелое поражение глаз с вторичной быстро прогрессирующей глаукомой;+
3) отсутствие аневризмы аорты и разболтанности суставов;+
4) снижение интеллекта.+
10. Гомоцистинурия - это:
1) наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в первую очередь метионина;+
2) наследственное заболевание, при котором из-за нарушения обмена галактозы в организме человека развиваются поражения печени, нервной системы и хрусталика глаза;
3) наследственное заболевание, связанное с дефектом аргиназы и проявляющееся высоким уровнем аргинина в крови;
4) редкое (орфанное) заболевание с аутосомно-доминантным типом наследования, обусловленное мутациями в гене фермента фумарилацетоацетазы (фумарилацетогидролазы (FAH)).
11. Гомоцистинурия вызвана дефицитом фермента:
1) галактозо-1-фосфат-уридилтрансферазы;
2) дигидрофолатредуктазы;
3) фенилаланин-гидроксилазы;
4) энзим цистатион-β-синтазы.+
12. Гомоцистинурия относится в МКБ-10 к классу:
1) M00-M99;
2) R00-R99;
3) U82-U85;
4) Е00-Е90.+
13. Для диагностики гомоцистинурии в качестве скринингового теста рекомендовано использовать:
1) клинический анализ крови;
2) метод тандемной масс-спектрометрии;
3) общий анализ мочи;
4) пробу на серосодержащие аминокислоты.+
14. Для диагностики классической гомоцистинурии рекомендовано использовать:
1) бактериологический метод;
2) копрограмму;
3) метод тандемной масс-спектрометрии;+
4) пробу на серосодержащие аминокислоты.
15. Для диагностики эктопии, сублюксации (или люксации) хрусталика необходима консультация:
1) кардиолога;
2) невролога;
3) офтальмолога;+
4) хирурга-ортопеда.
16. Для классической гомоцистинурии характерны:
1) низкий уровень метионина;
2) повышение уровня метионина;+
3) появление гомоцистина в сыворотке крови;+
4) снижение цистина в сыворотке крови и моче.+
17. Для подтверждения гомоцистинурии проводят тест с пиридоксином, где пациенту с начала дают:
1) 100мг пиридоксина;+
2) 250мг пиридоксина;
3) 300мг пиридоксина;
4) 500мг пиридоксина.
1) а, в, г;+
2) б, в, г;
3) б, г;
4) в, г.
1) а, б, в;
2) а, в, г;+
3) б, в, г;
4) в, г.
20. Концентрация гомоцистеина и гомоцистина в плазме должна быть определена у пациента, не получающего пиридоксин:
1) в течение 1 недели;
2) в течение 10 дней;
3) в течение 2 недель;+
4) в течение 5-7 дней.
21. Младенцы и дети дошкольного возраста не должны длительно получать пиридоксин в дозе:
1) более 100 мг/сут;
2) более 250 мг/сут;
3) более 300 мг/сут;+
4) более 500 мг/сут.
22. Назначение диетотерапии при установленном диагнозе гомоцистинурии, соответствует силе рекомендаций:
1) D;
2) А;
3) В;+
4) С.
23. Нарушение цикла преобразования метионина выражается в:
1) повышении уровня метионина и гомоцистеина в сыворотке крови;+
2) появлении гомоцистина;+
3) снижении уровня метионина и гомоцистеина в сыворотке крови;
4) уменьшении содержания цистина в крови.+
1) а, б, г;
2) а, в;+
3) а, в, г;
4) а, г.
1) а, б, в;+
2) а, в, г;
3) б, в;
4) б, в, г.
26. Патология сердечно-сосудистой системы при гомоцистинурии обусловлена развитием:
1) кардиомиопатии;
2) недостаточности митрального клапана;
3) стеноза аортального клапана;
4) тромбоэмболий в артериальных сосудах среднего и мелкого калибра.+
27. После установления гомоцистинурии у пациентов в возрасте до 1 года необходимо проводить мониторинг антропометрических показателей:
1) 1 раз в 3 месяца;
2) 1 раз в 6 месяцев;
3) 1 раз в год;
4) 1 раз в месяц.+
1) а, б, г;+
2) б, в;
3) б, в, г;
4) в, г.
29. При гомоцистинурии в анамнезе возможны указания на:
1) наличие сибсов с аналогичными клиническими признаками;+
2) наличие у близких родственников ранних инфарктов/инсультов;+
3) родственный брак;+
4) частые простудные заболевания.
1) а, б;
2) а, б, г;+
3) б, в, г;
4) б, г.
1) а, б, в;+
2) а, б, г;
3) а, в, г;
4) б, в.
1) а, б;
2) а, в, г;
3) б, г;+
4) в, г.
33. При классической гомоцистинурии характерны:
1) высокие показатели почечной экскреции гомоцистеина, гомоцистина;+
2) высокие показатели почечной экскреции метионина;+
3) низкие показатели почечной экскреции метионина;
4) низкие показатели почечной экскреции цистина.+
34. При лечении гомоцистинурии препаратами бетаина:
1) плазменный гомоцистеин преобразуется в метионин;+
2) повышается концентрация метионина в плазме;+
3) повышается концентрация плазменного гомоцистеина;
4) снижается концентрация плазменного гомоцистеина.+
35. При проведении теста с пиридоксином, после приема 100мг пиридоксина, повторяют измерение концентрации гомоцистина, гомоцистеина и метионина в крови:
1) через 12ч;
2) через 24ч;+
3) через 48ч;
4) через 6ч.
1) а, б, в;
2) а, в;+
3) а, г;
4) в, г.
37. Применение пиридоксина у детей 1 года жизни составляет:
1) 150-300 мг в день;
2) 200-500 мг в день;
3) 25-100 мг в день;+
4) 50-150 мг в день.
38. Результат теста с пиридоксином считается положительным, если отмечается снижение уровня гомоцистина, гомоцистеина и метионина в крови:
1) более 20%;
2) более 30%;+
3) менее 10%;
4) менее 20%.
39. Фенотипические черты больных гомоцистинурией:
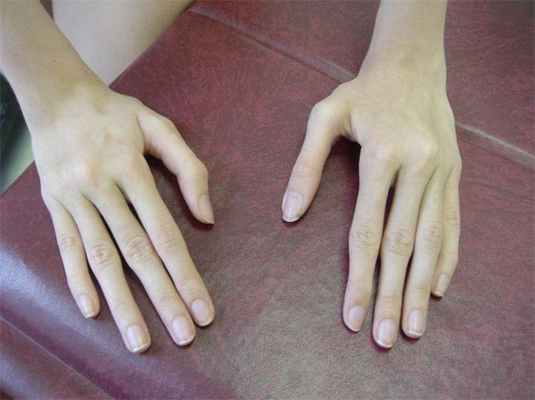
1) арахнодактилия кистей и стоп;+
2) астеническое телосложение;+
3) голубой цвет радужной оболочки;+
4) низкий рост.
40. Энзим цистатион-β-синтаза участвует в преобразовании метионина в:
1) аргинин;
2) лизин;
3) таурин;
4) цистеин.+
Генетика гомоцистинурии. Наследование
Стоимость исследования - 30000 руб
Срок исполнения - 30 рабочих дней
Гомоцистинурия представляет собой генетически обусловленное нарушение обмена серосодержащей аминокислоты метионина. В сыворотке крови и особенно моче людей, страдающих данным заболеванием, обнаруживается большое количество аминокислоты метионина, а также такого вещества, как гомоцистин (в норме не содержится в тканях и биологических жидкостях человека), и значительное снижение содержания аминокислоты цистина.
При гомоцистинурии первоначально возникает нарушение обмена серосодержащей аминокислоты метионина. В последующем происходит вовлечение в патологический процесс и других видов обмена веществ, что и является одной из причин развития всех проявлений данного заболевания, которые могут затрагивать совершенно различные органы и системы человеческого организма.
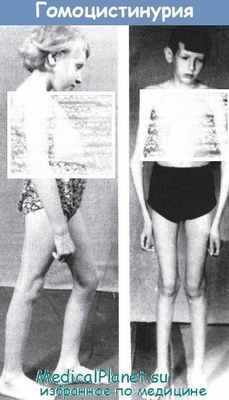
Для гомоцистинурии характерно наличие своеобразного комплекса симптомов, который включает в себя умственную отсталость с формированием симптомов поражения нервной системы (проявления зависят от уровня поражения нервной системы), эктопию хрусталиков, деформации скелета, тромбоэмболию и поражение сердечно-сосудистой системы. Люди, страдающие гомоцистинурией, как правило, имеют высокий рост, худощавое телосложение. Для них характерны длинные тонкие конечности, удлинение пальцев кистей и стоп, вальгусное положение коленных суставов, различные искривления позвоночника, воронкообразные или килевидные деформации грудной клетки, умеренно выраженный остеопороз костной системы. Из-за наличия остеопороза у лица с гомоцистинурией нередко возникают многочисленные переломы при незначительной травме. Наряду с этим, встречаются описания таких форм болезни, при которых изменения опорно-двигательного аппарата минимальны или отсутствуют вовсе.
Нервно-психическая деятельность при гомоцистинурии отличается инертностью (слабая переключаемость внимания и низкая работоспособность), некритичным отношением к своим возможностям, недостаточной осведомленностью об окружающей среде, в мышлении таких людей преобладает определенная направленность. Речь обычно состоит из коротких фраз, нередко с проявлениями дизлалии. Мимика при этом заболевании, как правило, слабо развита, иногда появляется немотивированная улыбка. Коэффициент интеллектуального развития соответствует при этом 32—72 единицам (при норме 85—115 единиц).
Гомоцистинурия имеет постоянно прогрессирующий характер течения. При этом у детей раннего возраста чаще всего не обнаруживается полного набора признаков болезни. Поражение глаз и изменения скелета формируются, как правило, не раньше 3—5-летнего возраста. Затем, по мере роста ребенка, происходит постепенное появление все новых признаков, и заболевание переходит в более тяжелую стадию — стадию декомпенсации, которая характеризуется развитием вторичной глаукомы, повторными инсультами и многими другими симптомами.
В связи с тем что все проявления гомоцистинурии очень напоминают таковые при синдроме Марфана (однотипные поражения глаз, опорно-двигательного аппарата), в некоторых случаях ставится неправильный диагноз. Гомоцистинурия отличается от синдрома Марфана типом наследования (при синдроме Марфана он является аутосомно-доминантным), более тяжелым поражением глаз из-за развития вторичной глаукомы, меньшим отставанием массы от роста ребенка, отсутствием аневризмы аорты и поражения клапанного аппарата сердца, меньшей степенью «разболтанности» суставов, плоскостопия, а также наличием остеопороза. Люди, страдающие гомоцистинурией, чаще блондины или светло-русые, с мягкими, слегка вьющимися волосами, голубым цветом глаз. Кроме того, характерные отличия, в виде появления определенных аминокислот и продуктов их обмена при гомоцистинурии, можно обнаружить при проведении лабораторных исследований сыворотки крови. При синдроме Марфана таких изменений состава крови не выявляется.
Гены CBS, MTHFR, MTR, MTRR, MMADHC участвуют в реакциях фолатного цикла и реметилировании гомоцистеина. Мутации в любом из них могут привести к гомоцистинурии.
гомоцистинурия
Гомоцистинурия – генетически обусловленная энзимопатия, характеризующаяся нарушением обмена незаменимой аминокислоты метионина, повышением уровня гомоцистина в биологических жидкостях и тканях, приводящим к повреждению органов и систем.
Начало изучению заболевания положено в 1962 г. Частота гомоцистинурии в популяции составляет
1 случай на 200 000 новорожденных. Течение гомоцистинурии сопровождается нервно-психическими нарушениями, глазной патологией, изменениями со стороны опорно-двигательного аппарата, склонностью к тромбоэмболиям.
Причины гомоцистинурии
Гомоцистинурия обусловлена аутосомно-рецессивным типом наследования. Аутосомно-рецессивный тип наследования тесно связан с гомоцистинурией. Здесь не имеет значения пол наследователя. Болезнь может проявляться при совпадении мутантных генов у ребенка от матери и отца. Если в роду возникла хотя бы одна мутация, то она будет передаваться из поколения в поколение до тех пор, пока не произойдет совпадение мутантных генов. На сегодняшний день известны 4 типа метаболических нарушений, которые могут лежать в основе патологии, в связи с чем выделяют следующие биохимические варианты заболевания:
Гомоцистинурия I – обусловлена отсутствием или снижением активности фермента цистатионин-бета-синтазы (классическая гомоцистинурия).
Гомоцистинурия II – обусловлена отсутствием или снижением активности фермента N5, N10-метилентетрагидрофолат-редуктазы.
Гомоцистинурия III – обусловлена низкой активностью фермента N5-метилентетрагидрофолата.
Гомоцистинурия IV – обусловлена отсутствием или снижением активности фермента гомоцистеин трансметилазы, вызванным дефектом синтеза метилкобаламина.
Симптомы гомоцистинурии
Проявления гомоцистинурии нарастают постепенно. Дети рождаются без каких-либо специфических отклонений. В течение первого года жизни развивается умеренно выраженная гипотрофия. Попытки устранить отставание в весе и росте за счет дополнительного введения в рацион белка в виде кефира или творога лишь усугубляют течение заболевания: нарастает дефицит массы тела, нарушается сон, ребенок становится раздражительным и плаксивым, отмечается позднее закрытие родничков, деформации конечностей, задержка психомоторного развития.
Обычно ярко выраженная клиника гомоцистинурии развивается в течение первых 10 лет жизни, однако часто диагноз становится очевидным уже в раннем детском возрасте. К этому времени у ребенка появляются высокоспецифичные глазные симптомы: подвывих хрусталиков, выраженная близорукость, дрожание радужки (иридодонез). Несколько позднее присоединяются астигматизм, глаукома, катаракта, отслойка сетчатки, атрофия зрительных нервов. Часто гомоцистинурии сопутствуют умственная отсталость, нарушения мышечного тонуса, гиперкинезы, судорожный синдром, поведенческие нарушения. Поражение опорно-двигательного аппарата включает килевидную деформацию грудной клетки, арахнодактилию, кифосколиоз, остеопороз, искривление голеней, полую стопу или плоскостопие, готическое нёбо. Практически половина пациентов с гомоцистинурией сталкивается с артериальными тромбозами (окклюзией церебральных, коронарных, почечных и периферических сосудов), а также венозными тромбозами (ТЭЛА).
Лица, страдающие гомоцистинурией, имеют определенные фенотипические черты: высокий рост, диспропорциональное телосложение (тонкие удлиненные конечности и укороченное туловище), голубые глаза, редкие светлые волосы. У них часто встречаются эритематозные пятна в области скуловых дуг, телеангиэктазии. Внешние проявления гомоцистинурии обладают определенным сходством с синдромом Марфана, однако для последнего не характерно снижение интеллекта и ряд других проявлений.

Диагностика гомоцистинурии
Пациенты с подозрением на гомоцистинурию должны направляться к медицинскому генетику для анализа генеалогических данных и проведения молекулярно-генетической диагностики. Диагноз устанавливается с помощью биохимического исследования крови и мочи: при гомоцистинурии в моче, плазме крови, ликворе обнаруживаются значительные количества гомоцистина, повышение содержания метионина при сниженном уровне цистина. В биоптатах кожи и печени выявляется специфический ферментативный дефект.
Рентгенологическое исследование трубчатых костей и позвоночника обнаруживает системный остеопороз. На ЭЭГ регистрируются нарушения биоэлектрической активности головного мозга, иногда пароксизмального характера. Консультация офтальмолога позволяет подтвердить характерные для гомоцистинурии нарушения со стороны зрительной системы. Также больные дети нуждаются в наблюдении и оценке развития со стороны педиатра, невролога, ортопеда, психиатра. Дифференциальная диагностика гомоцистинурии осуществляется с синдромом Марфана, последствиями родовой травмы и внутриутробных инфекций, другими энзимопатиями
Лечение гомоцистинурии
Лечебная тактика зависит от формы заболевания (В6-зависимой или В6-резистентной) и во многом схожа с лечением фенилкетонурии. При В6-резистентной форме гомоцистинурии необходимо соблюдение низкобелковой диеты, основанной на ограничении поступления в организм метионина. Рацион пациентов должен состоять, главным образом, из растительной пищи при исключении или значительном снижении употребления продуктов животного происхождения. Для возмещения потребности в незаменимых аминокислотах назначаются специальные аминокислотные смеси, лишенные метионина.
При В6-зависимой гомоцистинурии активность фермента удается активизировать назначением больших доз пиридоксина гидрохлорида.
При любых формах гомоцистинурии снижению уровня гомоцистина в биологических жидкостях способствует назначение фолиевой кислоты, бетаина. Для минимизации риска тромбозов показан постоянный прием ацетилсалициловой кислоты в низкой дозировке. По показаниям больным назначаются гепатопротекторы, ноотропы, препараты кальция, железа; проводятся курсы массажа, лазерной акупунктуры и рефлексотерапии, ЛФК.
Прогноз гомоцистинурии
Выявление заболевания на доклинической стадии, раннее начало лечения и соблюдение лечебной диеты позволяют отсрочить или предотвратить инвалидизирующие осложнения (интеллектуальные нарушения, параличи, атрофию зрительных нервов, легочное сердце, тяжелую артериальную гипертензию, инсульты, инфаркты внутренних органов и др.). В семьях, где есть носители гена гомоцистинурии, необходимо проведение инвазивной пренатальной диагностики с определением активности фермента в культуре клеток ворсин хориона или амниотической жидкости. В отношении детей с гомоцистинурией актуальны вопросы специализированного обучения, профессиональной ориентации, социальной адаптации, диспансерного наблюдения специалистов.
Консультация офтальмогенетика
Развитие молекулярной биологии и генетики даёт новые возможности для разработки методов диагностики и лечения заболеваний зрительного аппарата.
Технологии секвенирования ДНК позволяют во многих случаях быстро установить молекулярные механизмы конкретных заболеваний. Сведения о новых мутациях и их роли в этиологии и патогенезе расстройств органа зрения постоянно пополняются, в результате чего часто пересматривается классификация заболеваний. Использование в диагностике глазных болезней только клинических критериев осложняется большой вариабельностью симптоматических проявлений при том или ином заболевании. Всё большее число глазных заболеваний может быть верно и своевременно распознано только с помощью молекулярно-генетического анализа, и их применение уже становится стандартом в клинической науке и практике.
Новые возможности лечения заболеваний органа зрения, такие как генная терапия, также подчёркивают необходимость точной информации на молекулярном уровне. Таким образом, генетические исследования и основанная на них классификация многочисленных поражений органа зрения решает как диагностические, так и терапевтические задачи.
На данный момент изучены молекулярно-генетические основы при многочисленных поражениях органа зрения, включающих практически все описанные в настоящее время глазные заболевания и синдромы. Для большинства из них установлена связь с проявлениями заболевания. Накоплена клиническая, диагностическая и генетическая информация о более чем 650 наследуемых состояниях с глазными симптомами. Знание механизмов наследования позволяет решить многие проблемы, связанные с планированием беременности и рождением ребёнка без поражения глаз.
В нашей лаборатории вы можете получить консультацию специалиста офтальмолога-генетика, и по её итогам пройти генетическое обследование с выдачей заключения о характере патогенных вариантов в исследованных генах и дальнейшей тактике ведения пациента. Во многих случаях это позволит уточнить возможности проведения хирургического, консервативного или поддерживающего лечения, а также даст информацию о путях наследования поражения и возможности избежать передачу болезни потомству при планировании следующих/последующих беременностей.
Мы выполняем хромосомный микроматричный анализ, а также все виды исследований методом NGS: Клиническая «Офтальмо-панель», включающая анализ 443 генов, связанных с развитием наследственных заболеваний глаз, полноэкзомное и полногеномное секвенирование, секвенирование по Сэнгеру.
Обращение к офтальмологу-генетику позволит:
- Установить точный диагноз наследственной патологии органа зрения.
- Определить тип наследования заболевания.
- Оценить величину риска рождения ребёнка с наследственным заболеванием органов зрения и оказание помощи в принятии решения.
- В ряде случаев получить информацию о новых терапевтических опциях.
- При необходимости провести пренатальную (дородовую) диагностику, в том числе преимплантационное генетическое тестирование.
Симптомы, указывающие на возможность генетически обусловленных поражений органа зрения:
- Колобома верхнего века, колобома нижнего века, колобома радужки, колобома цилиарного тела, колобома сетчатки, колобома хориоидеи, колобома зрительного нерва
- Никталопия (ночная слепота), стационарная, прогрессирующая
- Врожденная катаракта
- Микрофтальм, микрофтальм с колобомой
- Микрокорнеа
- Макрокорнеа
- Задний эмбриотоксон
- Гипертелоризм
- Синофриз
- Врожденная гипертрофия пигментного эпителия сетчатки
- Врожденный нистагм, горизонтальный нистагм
- Атрофия зрительного нерва, частичная атрофия зрительного нерва
- Альбинизм, глазной альбинизм, глазо-кожный альбинизм
- Передний лентиконус, задний лентиконус
- Дистрофия/абиотрофия сетчатки, колбочко-палочковая дистрофия сетчатки, палочко-колбочковая дистрофия сетчатки, палочковая дистрофия сетчатки, колбочковая дистрофия сетчатки, центральная дистрофия сетчатки, смешанная дистрофия сетчатки, периферическая дистрофия сетчатки, пигментная дистрофия сетчатки (пигментная абиотрофия сетчатки, пигментный ретинит) вителлиформная дистрофия сетчатки (болезнь Беста), паттерн-дистрофии сетчатки, ювенильный ретиношизис
- Аниридия, врожденная аниридия, частичная аниридия, полная аниридия
- Гиперпигментация склеры
- Гипоплазия радужки, передние синехии
- Плоская роговица
- Эпикантус/эпикант
- Врожденный птоз верхнего века, птоз
- Частичная атрофия зрительного нерва, атрофия зрительного нерва, гипоплазия зрительного нерва
- Врожденная миопия
- Хориоретинальная атрофия/дисплазия/дистрофия
- Дистрофия роговицы, наследственная дистрофия роговицы (врожденная дистрофия роговицы), первичная дистрофия роговицы, врожденная эндотелиальная дистрофия роговицы, врожденная стромальная дистрофия роговицы
- Эктопия зрачка
- Наружная офтальмоплегия
- Гипоплазия фовеа
- Врожденная глаукома
- Иридогониодисгенез
- Микрокория
- Склерокорнеа
- Сферофакия
Заболевания, при которых установлены генетические причины поражения органа зрения
- Абеталипопротеинемия
- Аблефарон-макростомии синдром
- Акрофациальный дизостоз, тип Цинцинати
- Аденоматозный полипоз толстой кишки
- Адренолейкодистрофия
- Аакарди синдром
- Ал Каисси синдром
- Алажилля синдром
- Алландский глазной синдром, синдром Форсиуса-Эрикссона
- Синдром Альпорта
- Альстрома синдром
- Ангиопатия наследственная с нефропатией, аневризмой и мышечными спазмами
- Аниридия
- Ахроматопсия
- Алкаптонурия
- Дисгенез переднего сегмента, Аксенфельда-Ригера аномалия, Аксенфельда-Ригера синдром
- Аномалии лица, мозга и переднего сегмента глаза
- Синдром Аперта
- Первичная врожденная афакия
- Дисплазия удушающая торакальная
- Атаксия полинейропатия
- Атаксия с окуломоторной апраксией
- Атаксия телеангиэктазия
- Баллера-Герольда синдром
- Синдром Барайтсера-Винтера
- Горлина синдром (синдром базальноклеточных невусов, синдром Горлина-Гольтца)
- Атрофия зрительного нерва Бера
- Синдром Бьемонда
- Барде-Бидля синдром
- Кристаллическая дистрофия, корнеоретинальная дистрофия Биетти
- Блефароптоз, миопия, эктопия хрусталика
- Монохроматизм синих колбочек
- Брахио-окуло-фациальный синдром
- Синдром Элерса-Данло (Элерса-Данлоса)
- Брауна — Виолетта – Ван Лэре синдром
- Болезнь Канавана
- Церебро-окуло-фацио-скелетный синдром (COFS синдром, синдром Пена-Шокейра)
- Врожденная глаукома, ювенильная глаукома
- Врожденная миопия
- Центральная ареолярная хориоидальная атрофия
- Хориодеремия
- Болезнь Коактса
- Коэна синдром
- Колобома верхнего века, колобома нижнего века, колобома радужки, колобома цилиарного тела, колобома сетчатки, колобома хориоидеи, колобома зрительного нерва
- Дальтонизм, частичная цветовая слепота
- Дистрофия/абиотрофия сетчатки: колбочко-палочковая дистрофия сетчатки, палочко-колбочковая дистрофия сетчатки, палочковая дистрофия сетчатки, колбочковая дистрофия сетчатки, центральная дистрофия сетчатки, смешанная дистрофия сетчатки,
- Пигментная дистрофия сетчатки, пигментный ретинит
- Вителлиформная макулярная дистрофия, вителлиформная макулярная дегенерация, болезнь Беста
- Паттерн дистрофия макулы/ Паттерн дистрофия сетчатки
- Ювенильный ретиношизис
- Макулярная дистрофия, раннего начала
- Макулярная дистрофия северной Каролины
- Плоская роговица
- Дермоид роговицы, эпибульбарный дермоид
- Дистрофия роговицы Фукса, гранулярная дистрофия роговицы, пятнистая дистрофия роговицы, решетчатая дистрофия роговицы, дистрофия роговицы Месманна, задняя аморфная дистрофия роговицы, задняя полиморфная дистрофия роговицы, рецидивирующие эпителиальные эрозии, дистрофия роговицы Шнайдера, субэпителиальная муцинозная дистрофия роговицы, дистрофия роговицы Тиля-Бенке, Гранулярная дистрофия роговицы Авеллино
- Криптофтальм
- Болезнь Данона
- Сотовидная дистрофия сетчатки Дойна
- Синдром ретракции Дуэйна
- Врожденный дискератоз
- Эктопия зрачка
- Эктопия хрусталика
- Эндотелиальная дистрофия, гипоплазия радужки, врождённая катаракта, истончение стромы
- Эктодермальная дисплазия, эктродактилия, макулярная дистрофия
- Гипоплазия фовеа
- Эксфолиативная глаукома
- Наружная офтальмоплегия
- Болезнь Фабри
- Семейная экссудативная витреоретинопатия
- Фиброз экстраокулярных мышц
- Белоточечное глазное дно, белоточечная дистрофия сетчатки
- Врожденный амавроз Лебера, Амавроз Лебера
- Нейропатия Лебера, Атрофия зрительного нерва Лебера
- Галловей-Моват синдром, врожденный нефротический синдром
- Гапо синдром
- Гиллеспи синдром
- Гольденхара синдром
- Гольмана-Фавре синдром
- Синдром Горлина-Чаудри-Мосса
- Gyrate Atrophy
- Окуло-мандибуло-фациальный синдром (синдром Халлермана-Штрайфа)
- Синдром Харбояна
- Хеймлер синдром
- Синдром Германски-Пудлака
- Гомоцистинурия
- Болезнь Хантера
- Синдром Гурлера
- Синдром наследственной гиперферритинемии-катаракты
- Гипераксалурия
- Гипотрихоз с ювенильной макулярной дистрофией
- Синдром Блоха-Сульцбергера, или недержание пигмента
- Синдром Жалили/Джалили
- Жубера синдром
- Синдром Кабуки
- Окуло-церебро-фациальный синдром Кауфмана
- Синдром Кернса — Сейра
- Синдром Кенни-Коффи
- Врожденный кератит
- Кератоконус
- Кноблох синдром
- Болезнь Краббе
- Дисплазия Книста
- КИД-синдром, синдром кератит-ихтиоз-глухота
- Болезнь Норума
- Леопард синдром (LEOPARD синдром)
- Окуло-церебро-ренального синдрома Лоу
- Макрофтальм, колобоматозный с микрокорнеа
- Синдром Марфана
- Маринеску-Шегрена синдром
- Синдром Марото—Лами (мукополисахаридоз VI типа), синдром Моркио, (мукополисахаридоз IV типа), Санфилиппо синдром (мукополисахаридоз III типа)
- Синдром Маршалла
- Синдром Меккеля-Грубера, дизэнцефалия спланхнокистозная
- Мегалокорнеа, мегалокорнеа-эктопия хрусталика-сферофакия
- Синдром MELAS
- Анофтальм, анофтальмия
- Микрофтальмия, микрофтальм, микрофтальм с колобомой, микрофтальм с катарактой
- Врожденная миопия и глухота
- Нанофтальм, нанофтальм с пигментным ретинитом, нанофтальм с ретинопатией
- Нейрофиброматоз тип I, нейрофиброматоз тип II
- Нейрональный цероидный липофусциноз
- Врожденная стационарная ночная слепота
- Нунана синдром
- Болезнь Норри
- Глазо-зубо-пальцевой синдром
- Окуломоторная апроксия
- Болезнь Огуши
- Рото-лице-пальцевой (оро-фацио-дигитальный) синдром
- Нарушение биогенеза пероксисом, болезнь Цельвегера, Хеймлера синдром, неонатальная адренолейкодистрофия, синдром/болезнь Рефсума
- Первичное персистирующее гиперпластическое стекловидное тело (ППГСТ)
- Аномалия Петерса
- Синдром Пфайффера
- Пирсона синдром
- Пигментная паравенозная хориоретинальная атрофия
- Эластическая псевдоксантома
- Ретинобластома
- Рубинштейна-Тейби синдром
- Склерокорнеа
- Синдром Сенгера
- Сениора-Локена синдром
- Септооптическая дисплазия
- Синдром SHORT
- Синдром Смита-Лемли-Опица
- Синдром Смит—Магенис, синдром Смита—Магениса
- Сферофакия
- Болезнь Штаргардта, дистрофия сетчатки Штаргардта
- Стиклера синдром
- Болезнь Тея-Сакса
- Синдром Тричера Коллинз
- Туберозный склероз
- Ашера синдром (Ушера синдром)
- Болезнь Гиппеля — Линдау
- Синдром Ваарденбурга
- Вагнера синдром
- Синдром Уокера-Варбурга
- Синдром Уотсона
- Синдром Маркезани
- Синдром Вильямса, синдром Уильямса
- Болезнь Вильсона — Коновалова
- DIDMOAD-синдром, синдром Вольфрама
Медико-генетический центр и лаборатория
Широкий спектр услуг в области медицинской генетики

Мой геном - Планирование детей
Почему здоровым людям необходимо проходить генетический скрининг?
Более 5% детей рождаются с наследственными патологиями. Происходит это по причине того, что каждый второй родитель является носителем хотя бы одной патогенной мутации. Наличие двух таких генетических мутаций у ребёнка часто приводит к тяжёлым нарушениям физического и нервно-психического развития.
Вы можете быть абсолютно здоровы, но при этом передать наследственное заболевание будущему ребёнку. Зная генетические особенности обоих родителей, Вы можете спланировать успешную беременность и родить здорового ребенка.
Аутосомно-рецессивное наследование
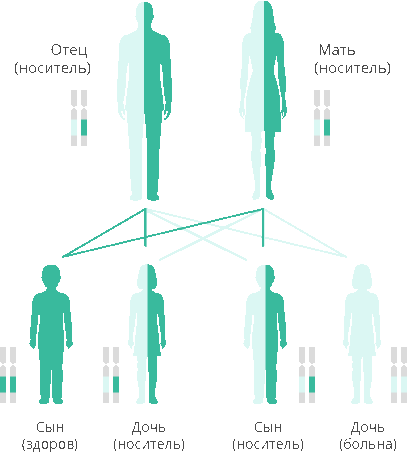
Большинство наследственных генетических заболеваний имеют аутосомно-рецессивный тип наследования.
Какие заболевания входят в тест ДНК "Планирование детей"?
Согласно рекомендациям Американской ассоциации акушеров и гинекологов по применению генетических тестов для оценки рисков рождения детей с наследственными патологиями:
1) Генетический тест должен включать в себя только те заболевания, которые существенно снижают качество и продолжительность жизни, приводят к физическим или ментальным нарушениям. Тест не должен включать в себя заболевания, которые проявляются во взрослом возрасте;
2) Информация о доступности генетического тестирования должна быть предоставлена каждой пациентке на приеме у любого акушера-гинеколога, в идеальном случае до зачатия;
3) Если человек оказался носителем мутации, ответственной за наследственное заболевание, то его партнеру рекомендуется пройти генетический тест;
4) Вспомогательные медицинские технологии могут снизить риск рождения больного ребенка. Такие варианты необходимо обсудить с родителями, являющимися носителями генетического заболевания.
Генетический тест включает 80 заболеваний и более 2000 мутаций.
Исследуемые заболевания:
- 1. Гепатолентикулярная дегенерация (Болезнь Вильсона-Коновалова)
- 2. GM1-ганглиозидоз, тип II
- 3. Алкаптонурия
- 4. Альфа-маннозидоз I, II
- 5. Альфа-талассемия
- 6. Аспартилглюкозаминурия
- 7. Атаксия-телеангиэктазия (синдром Луи-Бар)
- 8. Ахондрогенез, тип 1B
- 9. Ахроматопсия, тип 2, тип 3
- 10. Болезнь Кэнэвэн (болезнь Канавана)
- 11. Болезнь Тея-Сакса
- 12. Болезнь Унферрихта-Лундборга (Прогрессирующая миоклонус-эпилепсия, тип 1)
- 13. Болезнь Шарко-Мари-Тута, тип 4A
- 14. Восковидный липофусциноз нейронов, тип 5
- 15. Врожденная миотония, рецессивная форма, тип В 5, тип Фукуяма (тип А 4)
- 16. Врожденная мышечная дистрофия, тип В, 5
- 17. Врожденная мышечная дистрофия, тип Фукуяма, тип А, 4
- 18. Гликогеноз, тип II (Болезнь Помпе)
- 19. Глобоидно-клеточная лейкодистрофия (Болезнь Краббе)
- 20. Глутаровая ацидурия, тип 1
- 21. Гомоцистинурия
- 22. Дефицит митохондриального трифункционального белка
- 23. Диастрофическая дисплазия
- 24. Жёлтопятнистая абиотрофия сетчатки (Болезнь Штаргардта, тип 1; ювенильная форма макулярной дегенерации)
- 25. Конечностно-поясная мышечная дистрофия, тип 2A, тип 2D, тип 2E, тип C 1, тип C 5
- 26. Ларинго-онихо-кутанный синдром
- 27. Мандибулоакральная дисплазия
- 28. Мевалоновая ацидурия
- 29. Метахроматическая лейкодистрофия
- 30. Метилмалоновая ацидемия с полным отсутствием активности фермента
- 31. Миопатия Миоши, тип 3
- 32. Множественная эпифизарная дисплазия, тип 4
- 33. Муковисцидоз (кистозный фиброз)
- 34. Мукополисахаридоз IH (синдром Гурлер)
- 35. Мукополисахаридоз III A (синдром Санфилиппо А)
- 36. Недостаточность D-бифункционального белка пероксисом (синдром псевдоцелльвейгера)
- 37. Недостаточность ацил-KoA-дегидрогеназы среднецепочечных жирных кислот
- 38. Недостаточность биотинидазы
- 39. Недостаточность пальмитоилтрансферазы тип II
- 40. Нейродегенерация с отложением железа в мозге, тип 1 (Болезнь Галлервордена-Шпатца)
- 41. Нейрональный цероидный липофусциноз, тип 1 (болезнь Сантавуори-Халтиа)
- 42. Нейрональный цероидный липофусциноз, тип 2 (болезнь Янского-Бильшовского)
- 43. Нейрональный цероидный липофусциноз, тип 3 (болезнь Баттена-Фогта-Шпильмейера)
- 44. Нейрональный цероидный липофусциноз, тип 7, тип 8
- 45. Нейросенсорная тугоухость, тип 1A (аутосомно-рецессивная несиндромальная глухота)
- 46. Нейросенсорная тугоухость, тип 4
- 47. Нефропатический цистиноз
- 48. Нефротический синдром, тип 1
- 49. Пропионовая ацидемия
- 50. Рецессивный эритроцитоз
- 51. Семейная гиперинсулинемическая гипогликемия, тип 1 (незидиобластоз)
- 52. Семейная средиземноморская лихорадка (Периодическая болезнь)
- 53. Синдром Айкарди-Гутьереса, тип 2
- 54. Синдром Альстрёма
- 55. Синдром Андерманна (болезнь Шалерво, агенезия мозолистого тела с периферической нейропатией)
- 56. Синдром Барде-Бидля 1, 10
- 57. Синдром Гольдмана-Фавра
- 58. Синдром Коккейна, тип B (цереброокулофациоскелетный синдром (COFS) тип 1
- 59. Синдром Коэна
- 60. Синдром Криглера-Найяра, тип I
- 61. Синдром Лея (синдром Ли)
- 62. Синдром Пендреда
- 63. Синдром Смита-Лемли-Опица
- 64. Синдром Шегрена-Ларссона
- 65. Синдром нарушения гликозилирования, тип 1a, тип 1b
- 66. Синдром неймегеновского повреждения (синдром Ниймегена)
- 67. Спастическая атаксия Шарлевуа-Сагенэ
- 68. Тирозинемия, тип I
- 69. Фенилкетонурия
- 70. Цитруллинемия, тип I
Образец отчета



Исследуемые гены:
ATP7B, GLB1, HGD, MAN2B1, HBA2, AGA, ATM, SLC26A2, CNGA3, CNGB3, ASPA, HEXA, CSTB, GDAP1, CLN5, CLCN1, FKRP, FKTN, GAA, GALC, GCDH, CBS, HADHA, ABCA4, CAPN3, SGCA, SGCB, POMT1, LAMA3, LMNA, MVK, ARSA, MUT, ANO5, CFTR, IDUA, SGSH, HSD17B4, ACADM, BTD, CPT2, PANK2, PPT1, TPP1, CLN3, MFSD8, CLN8, GJB2, SLC26A4, CTNS, NPHS1, PCCA, PCCB, VHL, ABCC8, MEFV, RNASEH2B, ALMS1, SLC12A6, BBS1, BBS10, NR2E3, ERCC6, VPS13B, UGT1A1, SURF1, DHCR7, ALDH3A2, PMM2, MPI, NBN, SACS, FAH, PAH, ASS1
Читайте также: