Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
СодержаниеМукополисахаридоз: ферменты и болезньКакие бывают мукополисахаридозы?МПС и генетика«Люди-горгульи» и другие проявления болезниЛечение мукополисахаридозовСпецифическая терапияТрансплантация костного мозгаГенная терапияМукополисахаридоз в России
В этом году исполняется ровно 100 лет с тех пор, как канадец Чарльз Хантер составил первое описание мукополисахаридоза, выявленного у двух братьев. Сегодня известно, что это не одно, а целая группа заболеваний, очень редких. В мире проживает примерно 1,5 тысячи таких пациентов, около 200 из них живут в России. MedAboutMe выяснял, что такое мукополисахаридоз и какова судьбы людей, больных этим заболеванием.
Мукополисахаридоз: ферменты и болезнь
Немалая часть нашего организма состоит из соединительной ткани. Ее клетки участвуют в формировании каркаса и наружного покрова всех органов тела. В некоторых их них соединительная ткань составляет до 90% от их массы. Это хрящи, кости, жир, фасции, синовиальная жидкость, которая плещется в суставах, лимфа, склера и радужка глаза, микроглия, окружающая нейроны и др.
Без преувеличения, главным элементом соединительной ткани можно считать протеогликаны. Это соединения, которые содержат белковое ядро и связанные с ним многочисленные и разнообразные гликозаминогликаны (ГАГ). В разных тканях — разные протеогликаны. Они образуются, живут 7-10 дней и распадаются под действием ферментов — лизосомных гидролаз. Последние расщепляют именно ГАГ, причем каждому виду гликозаминогликанов соответствует свой личный фермент. Процесс образования (анаболизма) протеогликанов и распада (катаболизма) идет постоянно.
Что произойдет, если нужного фермента не окажется? Нерасщепленные или частично разрушенные протеогликаны станут накапливаться, откладываясь в соединительных тканях, пронизывающих все наше тело. Так и развиваются мупоколисахаридозы — разновидность лизосомных болезней накопления.
Какие бывают мукополисахаридозы?
На сегодняшний день известно о 8 типах заболевания, причем некоторые из них представлены в нескольких разновидностях. Обычно это связано с тем, что дефицит одного фермента может быть вызван разными мутациями — и, как следствие, проявления этих мутаций и срок жизни больных тоже могут различаться.
I тип объединяет три фенотипа: синдром Гурлер (МПС-IH), синдром Шейе (МПС-IS) и синдром Гурлер-Шейе (МПС-IH/S). Мутации, вызывающие синдром Гурлер и синдром Шейе, происходят в одном и том же гене. И иногда они развиваются одновременно. МПС I типа считается самой распространенной разновидностью болезни. Продолжительность жизни — от 6-10 (синдром Гурлер) до 30 лет (синдром Шейе). II тип — синдром Хантера (МПС-II). Это второй по частоте встречаемости вид мукополисахаридозов. Продолжительность жизни — 30-40 лет. III тип — синдром Санфилиппо, который подразделяется на 4 формы: A (МПС-III A), B (МПС-III B), C (МПС-III C) и D (МПС-III D). Это тоже разные мутации гена, который кодирует данный лизосомный фермент. Продолжительность жизни — до 20 лет. IV тип — синдром Моркио. Представлен двумя формами: A (МПС-IV A) и B (МПС-IV B). Продолжительность жизни — 20-35 лет. VI тип — синдром Марото-Лами (МПС-VI). Продолжительность жизни — 10-20 лет. VII тип — синдром Слая (МПС-VII). VIII тип — синдром Ди Ферранте (МПС-VIII) . XI тип — синдром дефицита гиалуронидазы, он же — синдром Натовича (МПС-IX).
Пятым типом раньше считался синдром Шейе.
Диагноз обычно ставят в детском возрасте.
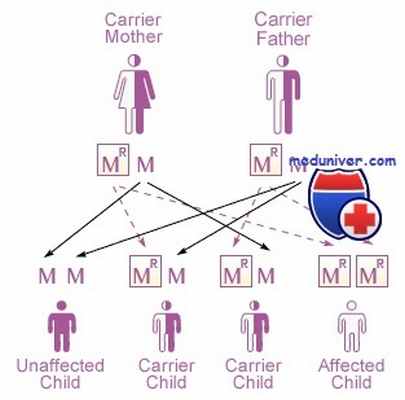
Мукополисахаридоз — это генетическое заболевание. Оно наследуется по аутосомно-рецессивному типу. Это значит, что мутация только на одной хромосомы из пары (а мы помним, что у человека 23 пары хромосом) не вызовет заболевания, потому что ген, расположенный на парной нормальной хромосоме будет исправно работать. Благодаря этому организм будет производить нужный фермент в требуемых количествах. А вот если человек получит на обеих хромосомах одинаковые мутации в области генов, кодирующих один и тот же лизосомный фермент — у него разовьется мукополисахаридоз. Чтобы такая ситуация сложилась, оба его родителя должны быть носителями бракованного гена. Такое совпадение случается очень редко — и поэтому мукополисахаридозы относятся к орфанным (редким) заболеваниям.
Из общего списка выбивается синдром Хантера (или Гунтера, в зависимости от перевода), МПС II типа. Именно это заболевание стало первым из открытых человечеством мукополисахаридозов. В отличие от генов, являющихся причиной других видов МПС, ген, вызывающий синдром Хантера, расположен на половой X-хромосоме. То есть его наследование сцеплено с полом, и болеют им в подавляющем большинстве случаев мальчики.
При других видах МПС, если болен один родитель, а второй здоров, ребенок получит одну хромосому с мутантным геном, а другую — такую же, но нормальную. Болезнь не разовьется. Но половые хромосомы Х и Y отличаются друг от друга. Мальчики получают Y-хромосому от отца и X-хромосому от матери. Если мать была обладателем хотя бы одной Х-хромосомы с мутантным геном (то есть даже сама не болела, была лишь носителем), и эта «неправильная» хромосома вошла в геном ребенка, то второй здоровой хромосомы ей в пару нет. Такой мальчик заболевает. «Люди-горгульи» и другие проявления болезни
Разные типы мукополисахаридоза имеют свои характерные особенности. Например, при МПС I, II и III типов развиваются тяжелые нарушения нервной системы, которые приводят к деменции (слабоумию). А есть и общие черты. Как мы помним, речь идет о заболеваниях, связанных с соединительной тканью. Поэтому неудивительно, что на их фоне развиваются тяжелые системные поражения костей, суставов, кожи.
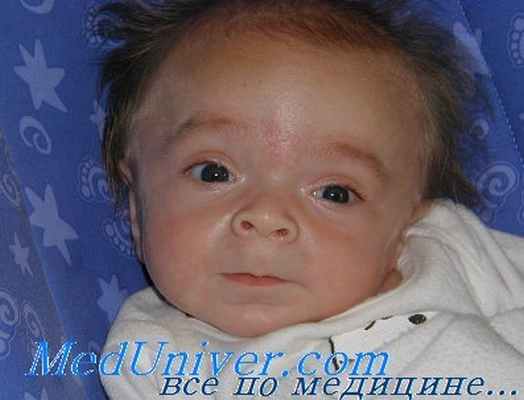
При этом может меняться форма головы, а при некоторых типах МПС уже с детства формируется специфическая внешность: приплюснутая переносица и нос с крупными ноздрями, широко посаженные глаза, толстые губы и приоткрытый рот, в целом грубые выразительные черты лица. Подметив сходство лиц таких пациентов с горгульями — каменными изображениями мифических существ характерного облика, врачи дали название данному состоянию — гаргоилизм (от французского слова gargouille).
Сегодня выделяют два основных фенотипа (внешние проявления) мукополисахаридоза:
Гурлер-подобный фенотип. В этом случае обычно развиваются деменция, гепатоспленомегалия (увеличение печени и селезенки) и гаргоилизм. Моркио-подобный фенотип. У таких пациентов нормальный интеллект и не настолько выражены повреждения скелета и связок, как при Гурлер-подобном фенотипе.
Все пациенты с МПС также страдают от задержки роста (нанизм) и диспропорционального развития скелета, нарушений зрения (помутнение роговицы и др.), слуха (тугоухость), заболеваний сердца и сосудов (аритмии, гипертрофия миокарда, заболевания клапанов сердца), болезней дыхательной системы.
Лечение мукополисахаридозов Специфическая терапия
Специфической называется терапия, мишенью которой являются непосредственные причины болезни. В данном случае это ферменты, которые не вырабатываются в достаточном количестве, и те нерасщепленные мукополисахариды, которые накапливаются и вызывают болезнь.
МПС-I — Альдуразим (ларонидаза); МПС-II — Элапраза (идурсульфаза); МПС-VI — Наглазим (галсульфаза).
Недостаток Элапразы заключается в том, что она не проникает сквозь гематоэнцефалический барьер, то есть не попадает в мозг. В 2015 году СМИ сообщали о начале лечения 31-летнего пациента с синдромом Хантера новым препаратом AGT-182 — комбинацией идурсульфазы и специально разработанного для этого антитела. Предполагалось, что это позволит лекарству проникнуть в мозг. Результаты эксперимента пока не опубликованы.
Специфических лекарств для лечения других типов МПС в России пока нет. Только симптоматическая терапия, направленная на уменьшение проявлений болезни, дает возможность таким больным прожить отведенный им срок.
Другой путь — субстратредуцирующая терапия, то есть угнетение выработки мукополисахаридов, избыток которых приводит к болезни. Пока такое лекарство разработано для МПС I, II и III типов (генистеин), но исследования проводились только на животных. В июне этого года ученые из Университета Манчестера сообщили о запуске проекта, в котором участвуют дети, больные МПС. Проект будет длиться год, и исследователи рассчитывают, что им удастся остановить разрушение центральной нервной системы при этом заболевании.
Трансплантация костного мозга
Ферментозаместительная терапия основана на введении экзогенных (то есть извне) синтетических ферментов. А трансплантация костного мозга (ТКМ) призвана заставить организм вырабатывать собственные, эндогенные ферменты при помощи пересаженных донорских стволовых клеток.
ТКМ — крайне сложная и очень дорогостоящая процедура. Она подходит далеко не всем пациентам и не со всеми типами МПС. Лучшие результаты были получены при пересадке костного мозга детям, страдающим МПС I типа, в возрасте 2 лет, то есть до того момента, пока поражение центральной нервной системы не стало слишком обширным. При попытках лечения методом ТКМ мукополисахаридозов II типа отмечается повышенная частота осложнений и летальность среди пациентов. Следует учитывать, что и сам процесс подбора неродственного донора возможен далеко не для всех пациентов.
Одним из самых значимых направлений в области разработки методов лечения МПС является генная терапия при помощи вирусных векторов — кольцевых ДНК, способных переносить нужный ген и встраивать его в заданный участок хромосомы. И уже есть первые положительные результаты.
В июле этого года ученые из Университета Миннесоты рассказали, как закапали в нос экспериментальным мышам препарат, содержащий вирус с геном, кодирующим лизосомный фермент альфа-L-идуронидазу — именно его так не хватает больным с МПС I типа. Введение препарата через нос позволило ему проникнуть через гематоэнцефалический барьер в мозг и защитить мозг от разрушительного действия накапливающихся мукополисахаридов. Теперь ученые ищут возможности повторить ту же самую процедуру с участием людей.
Мукополисахаридоз в России
Сегодня в России, по данным общественных организаций, занимающихся проблемой МПС, проживает около 200 пациентов с мукополисахаридозом. А по данным компании Санофи, в 2017 году диагноз МПС имели 98 россиян, причем 88 из них — это дети. Сегодня лечение получают только 77 из них: 52 человека находятся на пожизненной ферментозаместительной терапии, а 25 — прошли трансплантацию костного мозга (при синдроме Гурлер эта операция может проводиться только в возрасте от 2 до 4 лет). Из-за высокой стоимости лекарств, которые следует вводить еженедельно, расходы на это заболевание лидируют в бюджетах, выделенных на лечение орфанных болезней.
Например, для лечения людей с синдромом Хантера необходим препарат Элапраза. Стоимость одного флакона колеблется от 100 до 200 тысяч рублей. Для лечения одного пациента еженедельно требуется 300-500 тысяч рублей. И это пожизненная терапия. Должна быть.
В последние годы закупкой препаратов для пациентов с МПС занимались региональные власти. Нежелание тратить огромные деньги на отдельных пациентов с редкими болезнями приводило к бесконечным задержкам поставки лекарств — а это немедленно ухудшало состояние больных. Но недавно, в июне стало известно, что финансирование закупок препаратов для лечения МПС с будущего, 2018 года будет производиться из федерального бюджета. Это дает надежду многим из нынешних пациентов дожить до того момента, когда их болезнь можно будет вылечить.
Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
Генетика мукополисахаридозов. Наследование синдромов Гурлер, Шейе, Хантера
Мукополисахариды или гликозаминогликаны — полисахаридные цепи, синтезируемые клетками соединительной ткани как нормальный компонент многих тканей. Они состоят из большого числа дисахаридных блоков; отличительная черта конкретного гликозаминогликана — природа двух молекул сахаров.
Разложение этих макромолекул происходит в лизосоме и требует пошагового удаления блока моносахаридов в конце цепи с помощью фермента, специфичного для моносахарида и вовлеченной цепи. Таким образом, для разложения любого гликозаминогликана необходима серия ферментов, и один фермент часто участвует в распаде более чем одного гликозаминогликана.
Мукополисахаридозы — разнородная группа, включающая более дюжины болезней накопления, при которых в результате недостатка одного из ферментов, необходимого для их разложения, в лизосомах накапливаются мукополисахариды. При конкретных мукополисахаридозах могут накапливаться один или несколько гликозаминогликанов, если отсутствует фермент, необходимый для их распада.
Недеградировавшие гликозаминогликаны появляются в моче, где они могут быть обнаружены скрининговыми тестами.
Первые два описанных мукополисахаридоза — Х-сцепленный рецессивный синдром Хантера и более тяжелый аутосомно-рецессивный синдром Гурлер. Каждое из этих заболеваний сначала называли гаргоилизмом из-за грубости черт лица больных детей. Больные дети имеют скелетные аномалии и низкий рост, умственную задержку, а также другие аномалии.
Синдром Гурлер — следствие выраженной недостаточности а1-идуронидазы. Клинически отличное заболевание — синдром Шейе — первоначально считали вызванным мутацией в другом локусе, в основном из-за значительно более мягкого фенотипа. Тем не менее синдромы Шейе и Гурлер оказались аллельными, а мутации а1-идуронидазы, вызывающие синдром Шейе, связаны с более высокой остаточной активностью.
2. Шейе. Начало после 5 лет жизн, нормальный интеллект и продолжительность жизни, роговичные помутнения, клапанные пороки сердца. Дефект фермента: a-1-идуронидаза. Наследование: аутосомно-рецессивный. Очевидно, из-за аллелей, которые оставляют некоторую остаточную активность фермента.
3. Хантера. Гурлер-подобный синдром, но с медленным течением. Дефект фермента: Идуронатсульфатаза. Наследование: Х-сцепленный рецессивный. Мягкий фенотип с переменным поражением цнс.
Разные типы наследования аутосомного синдрома Гурлер и Х-сцепленного синдрома Хантера указывают, что они возникли вследствие мутаций в разных генах. Это различие также показано на клеточных культурах. Хотя фибробласты пациентов при обеих болезнях накапливают мукополисахариды в культуральной среде, накопление корректируется при совместном культивировании обоих типов клеток в одной культуральной посуде.
Коррекция происходит вследствие захвата фибробластами с недостаточностью a-L-идуронидазы при синдроме Гурлер нормальной a-L-идуронидазы, синтезированной фибробластами синдрома Хантера; в клетках с синдромом Хантера происходил обратный феномен. Этот простой эксперимент оказался красивой иллюстрацией того, что две болезни повреждают разные белки. Явление, когда продукт генома одного мутантного гена может скорректировать биохимический дефект у другого, называется генетической комплементацией, а исследования, используемые для обнаружения генетической комплементации, называют анализом комплементации.
Способность клетки усваивать недостающий лизосомный фермент из внеклеточной жидкости — один из механизмов коррекции биохимического дефекта при болезнях накопления после пересадки пациентам нормальных клеток (выделяющих фермент). Радикальный терапевтический эффект получен при лечении некоторых пациентов с мукополисахаридозами, включая синдром Гурлер, при пересадке костного мозга.
Способность клеток усваивать лизосомные ферменты из внеклеточной жидкости также дает обоснование для заместительной ферментной терапии при многих болезнях — стратегия, в значительном числе случаях оказавшаяся в высшей степени эффективной.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Только в 11% случаев педиатры смогли распознать мукополисахаридоз

Мукополисахаридоз – орфанное заболевание, которое возникает в результате мутации гена, ответственного за продукцию определенного фермента. О типах мукополисахаридоза, генетических аспектах, клинических проявлениях и актуальных методах лечения рассказала редакции к.м.н., доцент кафедры факультетской педиатрии РНИМУ им. Н.И. Пирогова, ведущий научный сотрудник отдела кардиоваскулярной патологии и орфанных болезней ЦКБ РАН Нато Джумберовна Вашакмадзе.
– Нато Джумберовна, в своей ежедневной практике врачи встречаются с широчайшим спектром нозологий. Как сохранить настороженность в отношении редких наследственных заболеваний?
– В первую очередь необходимо понимать, что большинство редких заболеваний имеют тяжелый характер и зачастую, сопровождаются жизнеугрожающим фактором. Об этом важно помнить и говорить, чтобы не терять бдительность, своевременно диагностировать заболевание и начинать лечение пациентов. В частности, мукополисахаридоз (МПС) – это хронически прогрессирующее заболевание, которое поражает многие органы: центральную нервную систему (у наиболее тяжелых пациентов), сердечно-сосудистую, дыхательную системы, костно-суставной аппарат, внутренние органы (печень, селезенка), также пациенты могут терять слух и зрение. Средняя продолжительность жизни пациента с МПС тяжелой степени составляет максимум 10 лет, при этом качество жизни ухудшается.
– Расскажите про типы МПС, чем они отличаются? Каково место МПС в структуре детской заболеваемости?
– Статистика говорит о том, что во всем мире у 1:100 000 новорожденных может быть мукополисахаридоз (МПС) I типа и примерно у 1:400 000 в России. МПС – крайне редкое наследственное заболевание, однако оно одно из самых дорогих в плане лечения пациентов и ухода за ними.
Существует 11 типов МПС, они различаются между собой степенью тяжести и характером клинических проявлений. Наиболее распространен МПС I типа (МПС I). Различают три клинических варианта его проявления: синдром Гурлер – одна из самых тяжелых форм МПС. В этом году будет 100 лет, как был описан синдром Гурлер. Он считается самым тяжелым, потому что дебют заболевания происходит в первые 3–4 месяца жизни ребенка и очень быстро прогрессирует. Также существует синдром Шейе (достаточно мягкая форма заболевания) и синдром Гурлер–Шейе (промежуточная степень).
– Выяснены ли до конца все генетические аспекты этого заболевания?
– МПС – это генетическое заболевание, которое имеет аутосомно-рецессивный тип наследования. Аутосомно-рецессивный тип наследования означает, что «дефектный» ген был унаследован от обоих родителей и при этом не содержится в половых (Х и Y) хромосомах, и вероятность того, что родится нездоровый ребенок с МПС, составляет 25%. Даже если родители не болеют, а являются носителями, то у них могут появиться как здоровые дети, так и нездоровые. Первые признаки заболевания могут проявиться как в младенчестве, так и в подростковом возрасте. Если в семье уже был случай МПС, родителям необходимо обязательно обратиться в центр планирования беременности для прогноза рождения здорового ребенка.
– На какие основные клинические проявления МПС I типа следует обратить внимание врачам первичного звена?
– К сожалению, только в 11% случаев педиатры смогли распознать МПС, хотя именно они видят ребенка минимум 11–12 раз за первый год жизни. Чаще всего выявляют заболевание неврологи, ортопеды и хирурги.
Также к симптомам МПС относятся увеличение печени (реже – селезенки) и костная деформация (грудопоясничный кифоз или контрактуры, которые не имеют никаких воспалительных проявлений). Может быть деформация суставов кистей рук, коленного или голеностопного сустава, деформация грудной клетки или позвоночника.
– Каковы актуальные подходы к диагностике и лечению?
– Ферментозаместительная терапия – пока единственный патогенетический метод лечения, который пациенты получают пожизненно. При МПС происходит мутация в гене, которая вызывает снижение или отсутствие активности определенного фермента. Например, для МПС-I это альфа-L-идуронидаза (IDUA). После многих исследований была выявлена оптимальная доза фермента, который используется в терапии МПС-I – 100 ед/кг еженедельно. Важно не снижать дозу и проводить лечение регулярно, нельзя также откладывать на длительный период старт терапии, это чревато неблагоприятными последствиями. По инструкции инфузия должна длиться 3–4 часа. Проводится процедура, как правило, в стационарах, дневных стационарах или амбулаторно.
Многие отечественные и зарубежные исследования доказали эффективность данной терапии. Но самый главный показатель – это результаты: выживаемость пациентов увеличилась на 10–15 лет, снижается прогрессирование осложнений сердечно-сосудистой, дыхательной систем, качество жизни пациентов заметно улучшается.
У нас был пациент в крайне тяжелом состоянии, которому поздно поставили диагноз, поздно начали лечение. Врачи говорили, что ему остался год. После ферментозаместительной терапии его состояние улучшилось, хотя много времени было потеряно. Прошло 8 лет, как я знаю этого мальчика, он все еще жив. Счастье видеть глаза мамы, когда она смотрит на своего единственного сына, которому становится лучше. Каждый день, когда ребенок просыпается – это счастье для родных.
– Почему ранняя диагностика и начало терапии очень важны для пациентов с МПС?
– Ранняя диагностика и лечение – залог лучшего качества жизни ребенка, хорошей выживаемости и улучшения его социальной адаптации. При своевременно начатой терапии патология разных систем не прогрессирует так быстро. Дети живут активной жизнью: посещают школу, обычную или специализированную, для них организуется очень много выездных мероприятий – летние лагеря, реабилитационные программы.
Важность своевременно начатого лечения мы можем наблюдать в своей практике. Мы наблюдаем случай, когда в одной семье болеют два ребенка. Старший ребенок начал получать лечение достаточно поздно, в возрасте 4 лет, у него болезнь достаточно сильно прогрессировала. А у младшего ребенка терапия была назначена в 6 месяцев – и сейчас никто не скажет, что у него есть такое тяжелое инвалидизирующее заболевание, как МПС: он пойдет в школу как обычный ребенок, может заниматься спортом.
– Насколько выстроена помощь больным с МПС в Москве и регионах?
– На данный момент пациенты могут получать лечение бесплатно, но какое-то время назад в связи с дороговизной препарата многие регионы не могли регулярно его поставлять, пациенты получали отказ в терапии, и лечение прерывалось. Сейчас федеральная программа по закупке препаратов решает очень много вопросов: автоматически закупается лекарство на конкретного ребенка с учетом его веса и роста, чтобы ребенок мог получать адекватную для него дозу препарата в течение года. Самое главное, что на данный момент требуется от регионов – это предоставлять правильные, актуальные антропометрические данные.
– Расскажите о работе лаборатории редких наследственных болезней у детей НЦЗД? Каковы основные направления работы?
– Существует несколько федеральных центров с лабораториями, которые занимаются орфанными болезнями. Специалисты, которые в них работают, имеют большой опыт ведения таких детей, хорошо понимают все нюансы. Очень важно, чтобы каждый ребенок получал максимально квалифицированную медицинскую помощь. И я считаю, что чем больше будет таких специализированных центров не только в крупных городах, но и в регионах, тем будет лучше, потому что пациентам из регионов очень сложно и дорого два раза в год на 2–3 недели приезжать на лечение в федеральные центры.
IX Международная студенческая научная конференция Студенческий научный форум - 2017

Лизосо́мные боле́зни накопле́ния (англ. Lysosomal Storage Diseases) — общее название группы весьма редких наследственных заболеваний, вызванных нарушением функции внутриклеточных органелл лизосом. Эти одномембранные органоиды, являются частью эндомембранной системы клетки и специализируются на внутриклеточном расщеплении веществ: гликогена, гликозаминогликанов, гликопротеинов и других. Лизосомные болезни накопления вызываются генетически обусловленным дефицитом ферментов лизосом, что приводит к накоплению макромолекул, являющихся субстратом этих ферментов, в различных органах и тканях организма
Мукополисахаридоз – это общее название довольно редко встречающихся генных заболеваний. Они напрямую связаны с патологией обмена кислых гликозаминогликанов, которые, в свою очередь, были вызваны дефицитным количеством лизосомных ферментов обмена гликозаминогликанов.
Виды заболевания
По характеру ферментативного дефекта (выраженных костных и обменных изменениях и психических патологиях) мукополисахаридоз бывает нескольких основных типов:
I тип, который имеет три основных фенотипа — это синдром Гурлер, синдром Гурлера-Шейе, синдром Шейе, которые характеризуются недостатком альфа-L-идуронидазов, быстрым развитием, наличием большого количества В и гепаритинсульфата в моче;
II тип — синдром Хантера, для которого характерны пигментный ретинит, тугоухость, большое количество гепаритинсульфата и хондроитинсульфата В в моче;
III тип — синдром Санфилиппо, для которого характерны тяжелое слабоумие и наличие большого количества гепаритинсульфата в моче;
IV тип — синдром Моркио, для которого характерны сильная деформация скелета, отсутствие помутнения роговицы, отсутствие снижения интеллекта и появления грубых черт лица;
V тип — синдром Шейе, для которого характерны помутнение роговицы и умеренно-выраженное искривление скелета;
VI тип — синдром Марото — Лами, для которого характерны замедления в росте, бочкообразная грудная клетка, грубые черты лица;
VII тип — синдром Слая дефицит р-глюкуронидазы, для которого характерен недостаток бета-глюкуронидазы.
Чем пациент становится старше, взрослеет, тем большие изменения скелета у него случаются, тем больше наблюдается патологий в разных системах и органах.
Причины мукополисахаридоза
Все формы и типы МПС относятся к категории наследственный патологий, передающихся аутосомно-рецессивным типом наследования. Мутационный ген представляет собой изменение структуры гена лизосомной альфа-ирунидазы, которая принимает непосредственное участие в метаболических превращениях глюкозаминогликанов.
Вследствие мутационного поражения лизосомной альфа-ирунидазы, происходит нарушение процесса внутрилизосомного распада дерматансульфата и избыточное его накопление в печеночной и селезеночной паренхиме, хрящевой ткани и надкостнице, нервных тканях и сосудистой стенке.
В результате прогрессирования отека мягкой оболочки мога развивается частичная окклюзия субарахноидального пространства, способствующая в свою очередь прогрессированию гидроцефалии. Причиной возникновения у ребенка признаков умственной отсталости является избыточное скопление ганглиозидов в нейронах.
Помимо выраженных нарушений метаболизма мукополисахаридов наблюдаеются принаки обменных нарушений белков,к которые проявляются в виде гипераминоацидурии.
Симптомы
Общие симптомы при мукополисахаридоз:
непропорционально малый рост и его задержка;
грубые черты лица: нависающий лоб, большой язык, гипертелоризм, деформация ушей и искривление зубов;
деформация грудной клетки;
выраженный кифоз грудного и поясничного отделов позвоночника;
ограничение подвижности суставов;
пупочные и паховые грыжи;
раннее окостенение затылочно-теменного шва;
диффузная мышечная гипотония;
общая двигательная заторможенность организма;
снижение уровня интеллекта;
Диагностика мукополисахаридоза
Мукополисахаридоз диагностируется у больного после полученных результатов исследований - клинического, генеалогического, биохимического, рентгено-функционального, молекулярно-генетического.
Врач назначает пациенту для обследования сдать анализы и пройти такие аппаратные осмотры, как:
исследование мочи на ГАГ;
определение активность лизосомальных гидролаз;
проведение рентгенологическое исследование;
проведение экскрецию гликозаминогликанов с мочой;
исследованиеактивность специфических ферментов;
исследование амниотическу жидкостью (повести антенатальную диагностику).
Лечение
В мировой врачебно практике при мукополисахаридозе есть несколько методик применяемого лечения:
физиотерапевтическое - электрофорез лидазы на область пораженных суставов, магнитотерапия, парафиновые аппликации, лазерная пунктура, ЛФК с воздействием на позвоночник и суставы, общий массаж, санация хронических очагов инфекции носоглотки и полости рта.
трансплантация стволовых клеток;
шунтирование при гидроцефалии;
операции карпального туннельного синдрома;
протезирование клапанов сердца.
Лечение мукополисахаридоза состоит в назначении таких гормональных препаратов, как:
АКТГ - с целью для уменьшить синтез мукополисахаридов;
Тем не менее, употребление гормональных препаратов дают всего лишь временные результаты.
Врач назначает больному также курс витамина А (в большом количестве), выписывает сердечные препараты.
Лечение мукополисахаридоза имеет симптоматический характер. Больного наблюдает целая группа специалистов. Это и ортопеды, которые занимаются ортопедической коррекцией патологий опорно-двигательного аппарата, и хирурги, которые вырезают грыжи посредством оперативного вмешательства, и педиатры, которые лечат постоянно проявляющиеся респираторные вирусные заболевания и следят за сердечно-сосудистой недостаточностью. Также к этому списку нужно прибавить оториноларингологов, которые лечат нарушения слуха, синуситы и хронические отиты, нейрохирургов, офтальмологов и невропатологов.
Профилактика мукополисахаридоза
К профилактическим средствам против мукополисахаридоза можно отнести пренатальную диагностику. Ее суть состоит в том, что врач определяет дефицит нужного фермента в амниотических клетках. Но, как таковых, профилактических рекомендаций против мукополисахаридоза, увы, не существует.
Заключение
Все формы заболевания характеризуются неблагоприятным прогнозом. Все дело в том, что с возрастом у пациента увеличиваются изменения скелета, а также нарушения различных систем и органов.
Без лечения у больных синдромом Гурлер наблюдается прогрессирующая физическая и психическая деградация и, как правило, гибель в возрасте до 10 лет. При синдроме Гурлер-Шейе симптомы болезни начинают проявляться позже и в менее тяжелой форме, а продолжительность жизни может превышать 20 лет. При синдроме Шейе больные доживают до зрелого возраста, у них может быть нормальный интеллект, но все же есть проблемы со здоровьем – такие как тугоподвижность суставов, нарушения зрения и слуха, пороки сердца.
Надежды семей, где есть больные синдромом Гурлер, связаны с аллогенной трансплантацией костного мозга или пуповинной крови. Если провести трансплантацию вовремя, то в большинстве случаев она заканчивается успешно. В разных странах известны примеры больных, которые через 10 и более лет после трансплантации ведут достаточно полноценную жизнь, включая самообслуживание в быту, обучение и общение со сверстниками.
Синдром Хантера типа В имеет более благоприятное течение и прогноз; при своевременном и регулярном лечении продолжительность жизни больных может достигать 50-60 лет. При тяжелых формах мукополисахаридоза II типа пациенты обычно погибают до 20 лет от сердечно-сосудистой недостаточности.
Мукополисахаридоз типа IV Средняя продолжительность жизни больных мукополисахаридозом – менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Мукополисахаридоз: короткая жизнь с надеждой на будущее

В этом году исполняется ровно 100 лет с тех пор, как канадец Чарльз Хантер составил первое описание мукополисахаридоза, выявленного у двух братьев. Сегодня известно, что это не одно, а целая группа заболеваний, очень редких. В мире проживает примерно 1,5 тысячи таких пациентов, около 200 из них живут в России. MedAboutMe выяснял, что такое мукополисахаридоз и какова судьбы людей, больных этим заболеванием.
Мукополисахаридоз: ферменты и болезнь
Немалая часть нашего организма состоит из соединительной ткани. Ее клетки участвуют в формировании каркаса и наружного покрова всех органов тела. В некоторых их них соединительная ткань составляет до 90% от их массы. Это хрящи, кости, жир, фасции, синовиальная жидкость, которая плещется в суставах, лимфа, склера и радужка глаза, микроглия, окружающая нейроны и др.
Без преувеличения, главным элементом соединительной ткани можно считать протеогликаны. Это соединения, которые содержат белковое ядро и связанные с ним многочисленные и разнообразные гликозаминогликаны (ГАГ). В разных тканях — разные протеогликаны. Они образуются, живут 7-10 дней и распадаются под действием ферментов — лизосомных гидролаз. Последние расщепляют именно ГАГ, причем каждому виду гликозаминогликанов соответствует свой личный фермент. Процесс образования (анаболизма) протеогликанов и распада (катаболизма) идет постоянно.
Что произойдет, если нужного фермента не окажется? Нерасщепленные или частично разрушенные протеогликаны станут накапливаться, откладываясь в соединительных тканях, пронизывающих все наше тело. Так и развиваются мупоколисахаридозы — разновидность лизосомных болезней накопления.
Какие бывают мукополисахаридозы?

На сегодняшний день известно о 8 типах заболевания, причем некоторые из них представлены в нескольких разновидностях. Обычно это связано с тем, что дефицит одного фермента может быть вызван разными мутациями — и, как следствие, проявления этих мутаций и срок жизни больных тоже могут различаться.
- I тип объединяет три фенотипа: синдром Гурлер (МПС-IH), синдром Шейе (МПС-IS) и синдром Гурлер-Шейе (МПС-IH/S). Мутации, вызывающие синдром Гурлер и синдром Шейе, происходят в одном и том же гене. И иногда они развиваются одновременно. МПС I типа считается самой распространенной разновидностью болезни. Продолжительность жизни — от 6-10 (синдром Гурлер) до 30 лет (синдром Шейе).
- II тип — синдром Хантера (МПС-II). Это второй по частоте встречаемости вид мукополисахаридозов. Продолжительность жизни — 30-40 лет.
- III тип — синдром Санфилиппо, который подразделяется на 4 формы: A (МПС-III A), B (МПС-III B), C (МПС-III C) и D (МПС-III D). Это тоже разные мутации гена, который кодирует данный лизосомный фермент. Продолжительность жизни — до 20 лет.
- IV тип — синдром Моркио. Представлен двумя формами: A (МПС-IV A) и B (МПС-IV B). Продолжительность жизни — 20-35 лет.
- VI тип — синдром Марото-Лами (МПС-VI). Продолжительность жизни — 10-20 лет.
- VII тип — синдром Слая (МПС-VII).
- VIII тип — синдром Ди Ферранте (МПС-VIII) .
- XI тип — синдром дефицита гиалуронидазы, он же — синдром Натовича (МПС-IX).
Пятым типом раньше считался синдром Шейе.
Диагноз обычно ставят в детском возрасте.
МПС и генетика

Мукополисахаридоз — это генетическое заболевание. Оно наследуется по аутосомно-рецессивному типу. Это значит, что мутация только на одной хромосомы из пары (а мы помним, что у человека 23 пары хромосом) не вызовет заболевания, потому что ген, расположенный на парной нормальной хромосоме будет исправно работать. Благодаря этому организм будет производить нужный фермент в требуемых количествах. А вот если человек получит на обеих хромосомах одинаковые мутации в области генов, кодирующих один и тот же лизосомный фермент — у него разовьется мукополисахаридоз. Чтобы такая ситуация сложилась, оба его родителя должны быть носителями бракованного гена. Такое совпадение случается очень редко — и поэтому мукополисахаридозы относятся к орфанным (редким) заболеваниям.
Из общего списка выбивается синдром Хантера (или Гунтера, в зависимости от перевода), МПС II типа. Именно это заболевание стало первым из открытых человечеством мукополисахаридозов. В отличие от генов, являющихся причиной других видов МПС, ген, вызывающий синдром Хантера, расположен на половой X-хромосоме. То есть его наследование сцеплено с полом, и болеют им в подавляющем большинстве случаев мальчики.
- При других видах МПС, если болен один родитель, а второй здоров, ребенок получит одну хромосому с мутантным геном, а другую — такую же, но нормальную. Болезнь не разовьется.
- Но половые хромосомы Х и Y отличаются друг от друга. Мальчики получают Y-хромосому от отца и X-хромосому от матери. Если мать была обладателем хотя бы одной Х-хромосомы с мутантным геном (то есть даже сама не болела, была лишь носителем), и эта «неправильная» хромосома вошла в геном ребенка, то второй здоровой хромосомы ей в пару нет. Такой мальчик заболевает.
«Люди-горгульи» и другие проявления болезни
Разные типы мукополисахаридоза имеют свои характерные особенности. Например, при МПС I, II и III типов развиваются тяжелые нарушения нервной системы, которые приводят к деменции (слабоумию). А есть и общие черты. Как мы помним, речь идет о заболеваниях, связанных с соединительной тканью. Поэтому неудивительно, что на их фоне развиваются тяжелые системные поражения костей, суставов, кожи.
При этом может меняться форма головы, а при некоторых типах МПС уже с детства формируется специфическая внешность: приплюснутая переносица и нос с крупными ноздрями, широко посаженные глаза, толстые губы и приоткрытый рот, в целом грубые выразительные черты лица. Подметив сходство лиц таких пациентов с горгульями — каменными изображениями мифических существ характерного облика, врачи дали название данному состоянию — гаргоилизм (от французского слова gargouille).
Сегодня выделяют два основных фенотипа (внешние проявления) мукополисахаридоза:
- Гурлер-подобный фенотип. В этом случае обычно развиваются деменция, гепатоспленомегалия (увеличение печени и селезенки) и гаргоилизм.
- Моркио-подобный фенотип. У таких пациентов нормальный интеллект и не настолько выражены повреждения скелета и связок, как при Гурлер-подобном фенотипе.
Все пациенты с МПС также страдают от задержки роста (нанизм) и диспропорционального развития скелета, нарушений зрения (помутнение роговицы и др.), слуха (тугоухость), заболеваний сердца и сосудов (аритмии, гипертрофия миокарда, заболевания клапанов сердца), болезней дыхательной системы.
Лечение мукополисахаридозов
Специфическая терапия

Специфической называется терапия, мишенью которой являются непосредственные причины болезни. В данном случае это ферменты, которые не вырабатываются в достаточном количестве, и те нерасщепленные мукополисахариды, которые накапливаются и вызывают болезнь.
- МПС-I — Альдуразим (ларонидаза);
- МПС-II — Элапраза (идурсульфаза);
- МПС-VI — Наглазим (галсульфаза).
Недостаток Элапразы заключается в том, что она не проникает сквозь гематоэнцефалический барьер, то есть не попадает в мозг. В 2015 году СМИ сообщали о начале лечения 31-летнего пациента с синдромом Хантера новым препаратом AGT-182 — комбинацией идурсульфазы и специально разработанного для этого антитела. Предполагалось, что это позволит лекарству проникнуть в мозг. Результаты эксперимента пока не опубликованы.
Специфических лекарств для лечения других типов МПС в России пока нет. Только симптоматическая терапия, направленная на уменьшение проявлений болезни, дает возможность таким больным прожить отведенный им срок.
Другой путь — субстратредуцирующая терапия, то есть угнетение выработки мукополисахаридов, избыток которых приводит к болезни. Пока такое лекарство разработано для МПС I, II и III типов (генистеин), но исследования проводились только на животных. В июне этого года ученые из Университета Манчестера сообщили о запуске проекта, в котором участвуют дети, больные МПС. Проект будет длиться год, и исследователи рассчитывают, что им удастся остановить разрушение центральной нервной системы при этом заболевании.
Трансплантация костного мозга
Ферментозаместительная терапия основана на введении экзогенных (то есть извне) синтетических ферментов. А трансплантация костного мозга (ТКМ) призвана заставить организм вырабатывать собственные, эндогенные ферменты при помощи пересаженных донорских стволовых клеток.
ТКМ — крайне сложная и очень дорогостоящая процедура. Она подходит далеко не всем пациентам и не со всеми типами МПС. Лучшие результаты были получены при пересадке костного мозга детям, страдающим МПС I типа, в возрасте 2 лет, то есть до того момента, пока поражение центральной нервной системы не стало слишком обширным. При попытках лечения методом ТКМ мукополисахаридозов II типа отмечается повышенная частота осложнений и летальность среди пациентов. Следует учитывать, что и сам процесс подбора неродственного донора возможен далеко не для всех пациентов.
Генная терапия

Одним из самых значимых направлений в области разработки методов лечения МПС является генная терапия при помощи вирусных векторов — кольцевых ДНК, способных переносить нужный ген и встраивать его в заданный участок хромосомы. И уже есть первые положительные результаты.
В июле этого года ученые из Университета Миннесоты рассказали, как закапали в нос экспериментальным мышам препарат, содержащий вирус с геном, кодирующим лизосомный фермент альфа-L-идуронидазу — именно его так не хватает больным с МПС I типа. Введение препарата через нос позволило ему проникнуть через гематоэнцефалический барьер в мозг и защитить мозг от разрушительного действия накапливающихся мукополисахаридов. Теперь ученые ищут возможности повторить ту же самую процедуру с участием людей.
Мукополисахаридоз в России
Сегодня в России, по данным общественных организаций, занимающихся проблемой МПС, проживает около 200 пациентов с мукополисахаридозом. А по данным компании Санофи, в 2017 году диагноз МПС имели 98 россиян, причем 88 из них — это дети. Сегодня лечение получают только 77 из них: 52 человека находятся на пожизненной ферментозаместительной терапии, а 25 — прошли трансплантацию костного мозга (при синдроме Гурлер эта операция может проводиться только в возрасте от 2 до 4 лет). Из-за высокой стоимости лекарств, которые следует вводить еженедельно, расходы на это заболевание лидируют в бюджетах, выделенных на лечение орфанных болезней.
Например, для лечения людей с синдромом Хантера необходим препарат Элапраза. Стоимость одного флакона колеблется от 100 до 200 тысяч рублей. Для лечения одного пациента еженедельно требуется 300-500 тысяч рублей. И это пожизненная терапия. Должна быть.
В последние годы закупкой препаратов для пациентов с МПС занимались региональные власти. Нежелание тратить огромные деньги на отдельных пациентов с редкими болезнями приводило к бесконечным задержкам поставки лекарств — а это немедленно ухудшало состояние больных. Но недавно, в июне стало известно, что финансирование закупок препаратов для лечения МПС с будущего, 2018 года будет производиться из федерального бюджета. Это дает надежду многим из нынешних пациентов дожить до того момента, когда их болезнь можно будет вылечить.

Ученые университета Дюка создали небольшой перечень показателей здоровья, обеспечивающих долголетие. Со статистической достоверностью можно утверждать, что люди, имеющие высокие показатели, могут рассчитывать прожить дольше среднего. Конечно, более точный результат можно получить, дополнив тест полным медицинским обследованием, однако и неформальный подход может помочь узнать многое о себе. Постарайтесь отвечать на вопросы как можно более честно и объективно.
Читайте также: