Генетика острой перемежающейся порфирии. Наследование
Добавил пользователь Алексей Ф. Обновлено: 07.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Порфирия: причины появления, симптомы, диагностика и способы лечения.
Определение
Порфирии (от греч. porphyreis – пурпурный) – это ряд заболеваний обмена веществ, при которых нарушается образование гема, представляющего собой комплексное соединение порфиринов с двухвалентным железом. В результате в организме происходит накопление порфиринов (пигментов, представляющих собой производные порфина) или их предшественников. К счастью, эти патологии встречаются относительно редко ‒ не более 7-12 случаев на 100 000 человек.
Причины появления порфирии
В подавляющем большинстве случаев причиной порфирий становятся генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в синтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается на фоне заболеваний печени (алкогольного гепатита, вирусного гепатита С) или длительной интоксикации тяжелыми металлами.
Наследование порфирий происходит по аутосомно-доминантному (наследуется одна нормальная и одна измененная копия гена, причем измененная копия доминирует и «подавляет» нормальную, в результате чего развивается генетическое заболевание) или аутосомно-рецессивному типу (болезнь Гюнтера). Синтез гема проходит 8 последовательных этапов, за каждый из которых отвечает конкретный фермент, кодируемый определенным геном. Соответственно, для каждой формы порфирии существует специфичный ферментативный дефект.
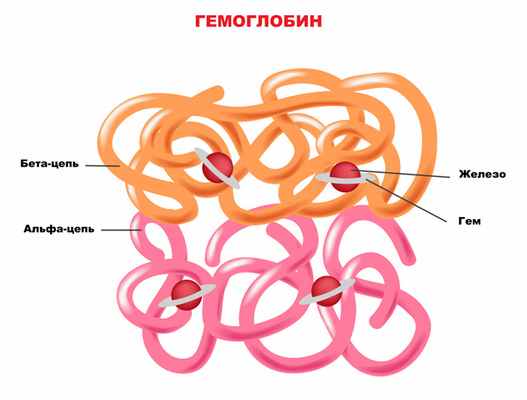
Наибольшее количество гемов образуется в печени и костном мозге. В печени гемы входят в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и «обезвреживании» различных ксенобиотиков (чужеродных химических веществ, попадающих в организм извне). В костном мозге гемы необходимы для синтеза гемоглобина.
Результатом снижения активности ферментов становится торможение синтеза гемов, что ведет к накоплению его токсичных промежуточных метаболитов.
Провоцирующими факторами развития порфириновой болезни становятся:
- избыточная инсоляция;
- недостаточное питание и скудный рацион;
- систематические стрессы;
- чрезмерное употребление алкоголя;
- вирусные и бактериальные инфекции;
- хронические интоксикации солями тяжелых металлов.
- Беременность.
В отдельных случаях манифестация патологического процесса может произойти на фоне приема некоторых лекарственных препаратов: антибиотиков, антиконвульсантов, нестероидных противовоспалительных средств, пероральных контрацептивов.
Классификация заболевания
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность.
Современная классификация порфирий весьма разветвленная:
I. Эритропоэтические порфирии.
- Эритропоэтическая уропорфирия (врожденная эритропоэтическая порфирия, или болезнь Гюнтера).
- Эритропоэтическая протопорфирия:
- манифестная форма;
- латентная форма.
- Эритропоэтическая копропорфирия.
- Пирролопорфирия (острая перемежающая порфирия, доминантная порфирия шведского типа):
- манифестная форма;
- латентная форма.
- Протокопропорфирия (варигатная, или смешанная порфирия, доминантная порфирия Южно-Африканского типа):
- кожная форма;
- острая форма без кожных проявлений;
- комбинированная форма с кожными и острыми проявлениями;
- латентная форма.
- Урокопропорфирия (поздняя кожная порфирия):
- манифестная приобретенная (симптоматическая) форма (развивается при интоксикациях гексахлорбензолом, опухолях печени и других патологических состояниях;
- манифестная наследственная форма (развивается у лиц с наследственной предрасположенностью);
- латентная наследственная форма (выявляется у родственников больных).
- Наследственная копропорфирия.
- Неклассифицированная печеночная порфирия.
- Гепатоэритропоэтическая порфирия.
- Неклассифицированная печеночная порфирия, протекающая с клиническим синдромом световой оспы.
- Патологические состояния, сопровождающиеся геморрагическим синдромом и нарушениями порфиринового обмена печеночного типа.
- Скрытое носительство мутантного гена. Симптоматика и биохимические изменения отсутствуют. Выявляется лишь снижение уровня эритроцитарной порфобилиногендезаминазы.
- Здоровое носительство дефектного гена. Наблюдаются минимальные биохимические изменения при отсутствии клинических проявлений.
- Латентная форма. Выявляется стертая симптоматика без типичных острых приступов болезни и выраженные нарушения порфиринового обмена.
- Манифестная форма. Отмечается яркая клиническая картина, протекающая в виде приступов. Приступы могут иметь постоянную выраженность или, наоборот, с каждым разом усиливаться. Может развиться острейшая форма (часто возникает при беременности) с грубыми неврологическими нарушениями и летальным исходом. Отмечаются выраженные биохимические отклонения порфиринового обмена.
- Исследование мочи. В общем анализе мочи выявляется повышенная относительная плотность, розовый или красно-бурый цвет мочи. Часто такую окраску моча приобретает только при длительном нахождении на свету. Существует специальный скрининговый тест с реактивом Эрлиха: при добавлении реактива к моче она в течение 5 минут становится красной. Этот анализ считается тестом первой линии, его обязательно следует проводить всем пациентам, как только у них заподозрена порфирия. При положительном результате выполняется количественное определение содержания в моче порфиринов, ДАЛК и ПБГ - при перемежающейся порфирии они повышены.
- Исследование крови. В биохимическом анализе часто обнаруживаются гипогликемия, снижение уровня натрия и осмолярности плазмы. В эритроцитах отмечается сниженная активность порфобилиногендезаминазы. При молекулярно-генетической диагностике выявляется мутация гена PBGD. Определение активности фермента и ДНК-диагностика в основном применяются при латентных и скрытых формах острой перемежающейся порфирии или при сомнительных результатах предыдущих анализов.
Симптомы порфирии
Сроки дебюта рассмотренных выше форм заболевания различны: эритропический тип проявляется в возрасте 3–5 лет, острый печеночный — в 14–16 лет, хронический печеночный — после 40 лет. После манифестации патологии пациенты сталкиваются со специфической симптоматикой.
При острых формах порфирии больные жалуются на сильные боли в животе, задержку стула, учащенное сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого).
Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью и снижением чувствительности по всему телу, прогрессирующей мышечной слабостью, судорожными припадками, различными психическими расстройствами (тревожностью, психомоторным возбуждением, бредом).
При поздней кожной форме на участках кожного покрова, подвергающихся постоянному воздействию солнечного света, формируется гиперпигментация, в результате чего кожа приобретает землистый или бронзовый оттенок.
Выраженными симптомами этой формы порфирии являются везикулезные и буллезные высыпания, которые покрыты корками, шелушение кожи, медленно заживающие эрозии, милиумы.
Кроме того, могут отмечаться явления гипертрихоза (избыточного оволосения) лобно-височной области лица, фотосенсибилизация.
При эритропоэтических порфириях наблюдаются признаки светочувствительности, причем даже более отчетливые, чем при поздней кожной порфирии. Пациент испытывает сильную боль, если на кожный покров попадают прямые солнечные лучи. Обширные эрозии оставляют после себя атрофичексие рубцы, что приводит к обезображиванию внешнего вида больного. В результате множественных атрофических рубцов на коже кистей рук развиваются контрактуры. Избыточное количество порфиринов приводит к тому, что зубная эмаль приобретает красновато-коричневый цвет (эритродонтия), моча становится красной или розовой.
Специфический признак эритропоэтической порфирии – утолщение, огрубение и уплотнение кожи периоральной и периорбитальной зон, крыльев и спинки носа, тыльной поверхности кистей.
Диагностика порфирии
При постановке диагноза учитывается наличие данного заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, беременность).
- Общеклинический № и биохимический анализ крови: АЛТ, АСТ, непрямой билирубин, сывороточное железо, ферритин.
Синонимы: Общий анализ крови, ОАК. Full blood count, FBC, Complete blood count (CBC) with differential white blood cell count (CBC with diff), Hemogram. Краткое описание исследования Клинический анализ крови: общий.
Молекулярно-генетическая диагностика острой перемежающейся порфирии Лучинина, Юлия Алексеевна

Лучинина, Юлия Алексеевна. Молекулярно-генетическая диагностика острой перемежающейся порфирии : диссертация . кандидата биологических наук : 14.01.21, 03.02.07 / Лучинина Юлия Алексеевна; [Место защиты: Гематол. науч. центр РАМН].- Москва, 2010.- 141 с.: ил. РГБ ОД, 61 11-3/129
Введение к работе
Актуальность проблемы
В ГНЦ РАМН работы по обследованию и лечению больных с нарушением порфиринового обмена ведутся с 1996г. За это время обследовано 638 человек с направительным диагнозом острая порфирия, среди которых выявлено 133 больных с острыми формами порфирий, а с диагнозом ОПП оказалось 102 человека. Такое доминирование острой перемежающейся порфирий, наиболее тяжелой и распространенной формы острых порфирий, определяет повышенную актуальность наиболее детального обследования пациентов с этим диагнозом.
Острая перемежающаяся порфирия вызывается частичным дефицитом фермента порфобилиногендезаминазы и клинически чаще всего проявляется после достижения пубертатного возраста. В мире известны единичные случаи заболевания детей, и все они связаны с гомозиготным носительством дефектного гена, в то время как взрослые за редчайшими исключениями являются гетерозиготными носителями.
Своевременная точная диагностика и адекватная терапия позволяют спасти подавляющее большинство больных. В период развернутых острых проявлений заболевания, как правило, удается установить правильный диагноз острой перемежающейся порфирий, основываясь на клинических признаках и биохимической диагностике. Что же касается асимптомных носителей, то для них даже биохимическая диагностика, основанная на измерении активности порфобилиногендезаминазы в эритроцитах, далеко не всегда дает однозначный ответ на вопрос о носительстве заболевания, поскольку диапазоны уровней активности фермента у таких пациентов перекрываются с нормальными значениями. Поэтому особое значение приобретает молекулярно-генетическое исследование, позволяющее выявлять латентных, асимптомных носителей дефектного гена, потенциально имеющих риск развития клинической стадии заболевания.
Острая перемежающаяся порфирия (ОПП), имея доминантный характер наследования, характеризуется невысокой пенетрантностью (по максимальным оценкам до 10-15%), свидетельствующей о том, что мутация в гене порфобилиногендезаминазы (ПБГД) является необходимым, но не достаточным условием клинического проявления болезни, основная тяжесть которого связана не с недостатком конечного продукта ферментативной реакции, катализируемой ПБГД, а с накоплением избытка токсичного субстрата-предшественника. Поскольку 50% -ное снижение активности ПБГД за счет мутантного аллеля не является достаточным основанием для образования патологического фенотипа, должны существовать какие-то дополнительные генетические факторы, предопределяющие клиническое проявление у гетерозиготных носителей мутантного гена. Интенсивные исследования в области молекулярной генетики острых порфирий ведутся во многих странах мира. Однако, многие авторы отмечают неполноценность современного медико-генетического консультирования острых порфирий из-за невозможности дифференцированного подхода к асимптомным носителям заболевания (особенно, детям и подросткам) и выделения среди них каких-либо групп риска. Это обусловлено отсутствием информации о дополнительных генетических факторах, сонаследующихся с мутантными генами и принципиально влияющих на патогенез заболевания. Такие факторы в настоящее время обнаружены только для двух
доминантно наследуемых форм порфирий, не относящихся к острым -
эритропоэтической протопорфирии и поздней кожной порфирии. Для острых порфирии, к которым относится ОПП, о таких факторах ничего не известно.
Цели исследования:
Определение спектра мутаций в гене ПБГД и характера их возникновения у больных ОПП из различных регионов РФ и стран СНГ, наблюдаемых в ГНЦ РАМН, с использованием эффективных систем генетической диагностики. Выявление дополнительных генетических факторов, предопределяющих клиническое проявление у гетерозиготных носителей мутантного гена.
Задачи исследования:
1) Поиск мутаций в гене ПБГД у больных ОПП, наблюдаемых в ГНЦ РАМН.
Разработка скрининговьгх методов тестирования наиболее часто встречающихся мутаций в гене ПБГД.
Выявление асимптомных носителей в семьях больных ОПП, у которых найдено мутационное нарушение в гене ПБГД.
Установление просхождения (полифилетическое или монофилетическое) наиболее распространенных мутаций в гене ПБГД у пациентов с ОПП при помощи анализа гаплотипов.
Поиск при помощи анализа полиморфных вариантов гена ПБГД функционально неполноценных аллелей этого гена, сочетание которых с мутантным аллелем может определять клиническое проявление ОПП.
Изучение возможного влияния на клинику ОПП сочетаний мутантного гена ПБГД с аллельными вариантами различных генов 1-й и 2-й фаз системы детоксикации: цитохромов Р450 (CYP1A1, CYP2E1), микросомальной эпоксидгидролазы (тЕРХНІ), глутатионтрансфераз (GSTM1, GSTT1), N-ацетилтрансферазы (NAT2).
Научная новизна:
Проведено исследование российских больных ОПП с целью выяснения распределения мутаций в гене ПБГД по типу и локализации в сравнении с другими популяциями. Мутационный анализ, проведенный для 75 больных, выявил 50 различных дефектов в гене ПБГД, 29 из которых ранее в мировой популяции не
встречались. Выявлен характер возникновения наиболее часто встречающихся мутаций и показано, что мутации 53delT и IVS13+2T—>G;+6(+G) имеют монофилетическое происхождение. Впервые изучена возможная ассоциация аллельных вариантов генов фазы 1: CYP1A1 (A2455G), CYP2E1 (G-1259C) и 4-х генов фазы 2: NAT2 (С481Т , G590A G857A), mEPHXl: Tyrll3His - 3-й экзон, Hisl39Arg - 4-й экзон, GSTM1 (Del), GSTT1 (Del) с клиническим проявлением ОПП.
Практическая ценность:
Установление носительства мутантного гена ПБГД у родственников больных ОПП и их своевременное информирование, позволяет избежать развития тяжелой клинической стадии болезни, что приводит к сокращению сроков их лечения, снижению инвалидизации и значительному улучшению качества жизни.
Анализ генетического полиморфизма гена N-ацетилтрансферазы и глутатионтрансферазы можно рекомендовать в качестве прогностического теста для оценки риска клинического проявления ОПП.
Положения, выносимые на защиту.
Мутационный анализ гена ПБГД, проведенный для 75 неродственных больных ОПП, выявил 50 различных генных дефектов, 29 из которых ранее в мировой популяции не встречались.
Выявление наиболее часто встречающихся мутаций в гене ПБГД и создание для них скрининговых систем тестирования позволяют в ряде случаев сократить время и денежные затраты, необходимые для проведения молекулярно-генетического обследования больных ОПП и их родственников.
Проведение генетической диагностики в семьях больных ОПП и выявление асимптомных носителей заболевания помогает избежать тяжелых клинических случаев, требующих дорогостоящего лечения и приводящих к инвалидизации больных.
Низкая пенетрантность ОПП (около 10%) свидетельствует о том, что мутация в гене ПБГД является необходимым, но не достаточным условием клинического
проявления болезни, которое в случае ОПП не зависит от сонаследования мутантного аллеля гена ПБГД с функционально неполноценным аллелем этого же гена дикого типа.
Исследование полиморфизмов различных генов системы детоксикапии показало неслучайную ассоциацию между клиническим проявлением заболевания и генотипом N/N по полиморфизмам гена NAT2.
Несмотря на отсутствие статистически достоверных отличий, можно говорить об определенном влиянии полиморфных вариантов генов GSTT и GSTM на клиническое проявление заболевания.
По теме диссертации опубликовано 6 печатных работ.
Апробация работы
Диссертационная работа апробирована 5 июля 2010 года на совместном заседании проблемных комиссий «Гемопоэз, молекулярная биология, биотехнология, иммуногематология; гемобластозы и депрессии кроветворения» и «Опухоли лимфатической системы, патология красной крови, порфирии» Учреждения Российской академии медицинских наук Гематологический научный центр РАМН.
Основные положения работы доложены на Международной конференции по генетике человека (Прага, 2005г.) и Всероссийской научно-практической конференции «Молекулярные методы диагностики моногенных заболеваний: возможности и перспективы» (Москва, 2006г.).
Острые порфирии
Острые порфирии обусловлены дефицитом некоторых ферментов биосинтеза гема, приводящим к накоплению предшественников гема, которые вызывают периодические приступы боли в животе и неврологические симптомы. Приступы провоцируются некоторыми медикаментозными препаратами и другими факторами. Диагноз основывается на повышенных уровнях предшественников порфиринов – дельта-аминолевулиновой кислоты и порфобилиногена в моче во время приступов. Приступы купируют введением глюкозы или (в более тяжелых случаях) внутривенным введением гема. При необходимости проводят симптоматическую терапию, включая применение анальгетиков.
Острые порфирии включают, в порядке уменьшения распространенности:
Острая перемежающаяся порфирия (ОПП)
Смешанная порфирия (СП)
Наследственная копропорфирия (НКП)
Порфирия вследствие дефицита дегидратазы дельта-аминолевулиновой кислоты (ALAD) (крайне редкая)
При смешанной порфирии и наследственной копропорфирии, независимо от наличия нейровисцеральных симптомов, могут развиваться буллезные поражения кожи, особенно на руках, предплечьях, лице, шее или других областях кожи, подвергающихся воздействию солнечного света.
У гетерозигот острые порфирии до пубертатного возраста проявляются редко, а позднее – только у 2-4% носителей дефектов. У гомозигот и сложных гетерозигот заболевания часто проявляются более тяжелыми симптомами и, как правило, в детском возрасте.
Провоцирующие факторы
Эффект многочисленных провоцирующих факторов обычно связан со стимуляцией биосинтеза гема в той степени, которая превышает возможности дефектного фермента. В результате накапливаются предшественники порфирина – порфобилиноген (ПБГ) и дельта-аминолевулиновая кислота (АЛК), а в случае ALAD-дефицитной порфирии – только АЛК.
Возможной причиной приступа бывают сразу несколько факторов, которые порой трудно идентифицировать. Провоцирующие факторы включают:
Гормональные изменения у женщин
Низкокалорийная, низкоуглеводная диета
Воздействие органических растворителей
Инфекции и другие болезни
Важную роль играют гормональные факторы. У женщин приступы возникают чаще, чем у мужчин, особенно в периоды гормональных сдвигов (в лютеиновую фазу менструального цикла, при использовании пероральных контрацептивов, на ранних сроках беременности или в раннем послеродовом периоде). Тем не менее беременность не противопоказана.
При смешанной порфирии и наследственной копропорфирии воздействие солнечного света вызывает кожные проявления.
Симптомы и признаки острых порфирий
Для острых порфирий характерны симптомы и признаки поражения нервной системы, боли в животе или то и другое вместе (нейровисцеральные проявления). Приступы развиваются в течение часов или дней и могут продолжаться несколько недель. Большинство носителей дефектного гена за всю жизнь испытывают всего несколько приступов или вообще не испытывают их. У других симптомы рецидивируют. У женщин рецидивирующие приступы часто приурочены к лютеиновой фазе менструального цикла.
Приступ острой порфирии
Симптоматика обострения неспецифична и может быть схожа с таковыми многих других заболеваний. Запор, утомляемость, изменения психического состояния (часто описываются как «затуманивание») и бессонница обычно предшествуют острому приступу. Наиболее частые симптомы – боль в животе и рвота. Боль бывает мучительной и не соответствует напряжению мышц брюшной стенки или другим данным физикального обследования. Абдоминальные проявления могут возникать в связи с раздражением висцеральных нервов. Обычно воспаление отсутствует, живот остается мягким; признаки раздражения брюшины отсутствуют. Тем не менее, у небольшого количества пациентов с острыми печеночными порфириями также развивается острый панкреатит Острый панкреатит Острый панкреатит – это острое воспаление поджелудочной железы (иногда и прилегающих тканей). Наиболее распространенные триггерные механизмы – камни в желчных протоках и злоупотребление алкоголем. Прочитайте дополнительные сведенияПоражаться могут все отделы периферической нервной системы и центральной нервной системы. Для тяжелых и длительных приступов характерна моторная нейропатия. Вначале обычно поражаются мотонейроны конечностей (приводя к слабости рук и ног), но в процесс могут вовлекаться любые мотонейроны и черепные нервы; возможно развитие тетраплегии. Бульбарные поражения приводят к дыхательной недостаточности.
Повреждения центральной нервной системы могут проявляться судорогами или психическими отклонениями (например, апатией, депрессией, возбуждением и даже явным психозом и галлюцинациями). Судороги, психотическое поведение и галлюцинации могут быть обусловлены или усиливаться под влиянием гипонатриемии или гипомагниемии, которые сопровождаются сердечными аритмиями. Гипонатриемия может развиться во время приступа из-за чрезмерного выделения вазопрессина (антидиуретический гормон [АДГ]) и/или внутривенного введения гипотонических растворов (5% или 10% глюкоза в воде) - стандартной терапии приступов. У некоторых пациентов развиваются признаки синдрома задней обратимой энцефалопатии (PRES), характеризующегося головной болью, изменением психического статуса, судорогами и потерей зрения, предположительно из-за отека в области задних затылочных и теменных долей.
Избыток катехоламинов обычно вызывает беспокойство и тахикардию. В редких случаях катехоламиновые аритмии являются причиной внезапной смерти. Лабильная гипертензия с транзиторными подъемами артериального давления, если ее не лечить, вызывает изменения сосудов, ведущие к необратимой гипертонии. В основе хронического заболевания почек при острой порфирии лежат многие факторы; главным среди них является, вероятно, гипертензия (потенциально переходящая в хроническую гипертензию). Тем не менее у пациентов с острой перемежающейся порфирией, у которых развилась хроническая болезнь почек, были обнаружены генетические факторы, особенно генетические вариации пептидного переносчика 2 (PEPT2), который кодирует пептиды и переносчики аминокислот, способные влиять на реабсорбцию АЛК в проксимальных канальцах. В частности, было обнаружено, что носители варианта с более высокой аффинностью (PEPT2*1*1) имеют более тяжелую почечную дисфункцию, чем носители вариантов с более низкой аффинностью.
Подострые или хронические симптомы
У некоторых больных симптомы сохраняются более длительное время, но выражены слабее (например, запоры, утомляемость, головные боли, боли в пояснице или бедрах, парестезии, тахикардия, одышка, бессонница, депрессия, тревожность или другие нарушения настроения, судороги). Хронические симптомы между приступами, вероятно, возникают у многих пациентов, особенно у тех, кто пережил более одного острого приступа, наиболее частым симптомом является боль. Многие пациенты имели ежедневные проявления болезни.
Кожные проявления при смешанной порфирий и наследственной копропорфирии
Даже в отсутствие нейровисцеральных симптомов кожа становится легко ранимой и на открытых местах тела появляются буллезные высыпания. Больные часто не знают, что им нельзя находиться на солнце. Кожные проявления идентичны наблюдаемым при поздней кожной порфирии Поздняя кожная порфирия Поздняя кожная порфирия (ПКП) – сравнительно частая печеночная порфирия, при которой поражается преимущественно кожа. Также нередко имеют место заболевания печени. ПКП связана с приобретенной. Прочитайте дополнительные сведенияПоздние проявления острых порфирий
Нарушения моторики во время острых приступов могут стать причиной постоянной мышечной слабости и атрофии мышц, выявляемых между приступами. Во второй половине жизни у больных с острой интермиттирующей порфирией и, возможно, со смашанной порфирией и врожденной копропорфирией, особенно после перенесенных приступов, возрастает частота развития цирроза, печеночно-клеточного рака, системной артериальной гипертонии, почечной недостаточности.
Диагностика острых порфирий
Анализ мочи на порфобилиноген (ПБГ)
При положительных результатах – количест-венное определение АЛК и ПБГ в моче
Для подтверждения острой интермиттирующей порфирии измеряют активность деаминазы ПБГ в эритроцитах
При необходимости выяснить тип заболевания необходим генетический анализ
Острый приступ
Диагноз часто бывает ошибочным, так как острый приступ имитирует состояние острого живота Острая боль в животе Боль в животе – часто встречающийся симптом, который не всегда имеет очень важное значение. Однако острая и выраженная боль практически всегда служит признаком заболевания органов брюшной полости. Прочитайте дополнительные сведения (что подчас приводит к ненужной хирургической операции) или нервное или психическое заболевание. Приступ порфирии следует подозревать у больных, у которых ранее выявлено носительство дефектного гена, или у тех, у кого имеются указания на порфирию в семейном анамнезе. Однако даже в известных случаях носительства дефектного гена необходимо оценить возможность других причин острого приступа.
Основным признаком является красный или красно-коричневый цвет мочи, которого не было до начала приступа и который часто присутствует при полноценном приступе. Поэтому исследовать мочу нужно у всех пациентов, жалующихся на боль в животе (не имеющей явной причины), особенно при наличии запора, рвоты, тахикардии, мышечной слабости, бульбарных симптомов или психических отклонений.
При подозрении на порфирию содержание ПБГ в моче определяют быстрыми качественными или полуколичественными методами. Положительные результаты анализа или убедительная клиническая картина требуют количественного определения АЛК и ПБГ в моче, измерения уровня креатинина (лучше в тех же пробах мочи, что были исследованы ранее). Содержание ПБГ и АЛК, нормализованное к концентрации креатинина в моче, превышающее норму > чем в 5 раз, указывает на острый приступ порфирии, если только пациент не является носителем дефектного гена, у которого столь же высокая экскреция предшественников порфирина имела место и в латентной фазе заболевания.
Если соотношение ПБГ/креатинин и АЛК/креатинин в пределах нормы, следует подумать о другом диагнозе. Целесообразно провести измерение общего уровня порфиринов в моче и оценку профилей этих порфиринов методом высокоэффективной жидкостной хроматографии. Повышенное содержание АЛК и копропорфирина в моче при нормальном или слегка повышенном уровне ПБГ указывает на свинцовое отравление Интоксикация свинцом При отравлении свинцом симптомы незначительны в начале, однако в дальнейшем могут развиться острая энцефалопатия и необратимые изменения внутренних органов, обычно приводящие к когнитивным расстройствам. Прочитайте дополнительные сведения , дегидратаза дельта-аминолевулиново (ALAD)-дефицитную порфирию, или наследственную тирозинемию типа 1. Анализ суточной мочи в таких случаях не требуется. Вместо этого анализируют случайные порции мочи с поправкой на разведение по уровню креатинина.
Определение типа острой порфирии
Поскольку терапия при острой порфирии любого типа одинакова, выяснение типа заболевания имеет значение в основном для обнаружения носителей дефектного гена среди родственников больного. Если в семейном анамнезе уже имеются данные о типе порфирии и мутации, диагноз очевиден, но его можно подтвердить результатами генетического анализа.
Активность ферментов ALAD и PBG дезаминазы в эритроцитах легко измерить, что может быть полезно для установления диагноза ALAD-дефицитной порфирии и острой перемежающейся порфирии, соответственно. При порфирии, обусловленной дефицитом дегидратазы дельта-аминолевулиновой кислоты (ALAD), активность ALAD в эритроцитах сильно снижена (
Если же в семейном анамнезе нет указаний на диагноз, формы острой порфирии различают по накоплению типичных соединений в плазме и их экскреции с мочой и калом. При повышенных уровнях АЛК и ПБГ в моче определяют содержание порфиринов в кале. Для острой перемежающейся порфирии характерен нормальный или лишь слегка повышенный их уровень в кале, тогда как для наследственной копропорфирии и смешанной порфирии – высокий. В латентной фазе заболевания эти маркеры часто отсутствуют. Флуоресценцию при возбуждении плазмы светом из полосы Соре (~410 нм) можно использовать для дифференциации наследственной копропорфирии и смешанной порфирии, пиковые эмиссии которых различаются.
Обследования членов семьи при острой порфирии
Дети носителя гена аутосомно-доминантной формы острой порфирии (острая интермиттирующая порфирия, врожденная порфирия, смешанная порфирия) имеют 50% риск наследования заболевания. В отличие от этого, дети пациентов с ALAD-дефицитной порфирией (аутосомно-рецессивное наследование) являются облигатными носителями, но развитие клинически проявляющегося заболевания у них очень маловероятно. Поскольку терапевтические рекомендации после раннего установления диагноза снижают риск проявления болезни, детей в пораженных семьях необходимо обследовать до начала полового созревания. Если мутация известна, у ребенка проводят генетический анализ. Если нет, то измеряют соответствующие уровни фермента в эритроцитах или лейкоцитах. Тем не менее для постановки окончательного диагноза предпочтительным является генетическое тестирование, поскольку измерение ферментативной активности в эритроцитах подвержено вариациям в том, как образцы обрабатываются и отсылаются; существует перекрытие показателей активности дезаминазы ПБГ у лиц без порфирии и пациентов с ОПП, около 5% пациентов с ОПП имеют нормальную активность дезаминазы ПБГ в эритроцитах. Генетические исследования проводят и для внутриутробной диагностики (путем амниоцентеза Амниоцентез Генетическая оценка является частью рутинного пренатального наблюдения, в идеале выполняется до зачатия. Объем исследований, включенных в генетическую оценку, зависит от того, как женщина оценивает. Прочитайте дополнительные сведенияПрогноз при острых порфириях
Прогресс медицины и методов самопомощи улучшают прогноз больных с симптомами острой порфирии. Однако у некоторых из них все еще сохраняются частые кризы или развиваются постоянные параличи и почечная недостаточность. Кроме того, потребность в опиоидных анальгетиках может привести к распространению опиоидной зависимости.
Подводные камни в диагностике острой перемежающейся порфирии: описание клинического случая

Острая перемежающаяся порфирия — это редкое заболевание с аутосомно-доминантным типом наследования, вызванное дефицитом фермента гидроксиметилбилан-синтазы. Распознавание острых нейровисцеральных атак может быть затруднено из-за неспецифического характера симптомов.
Описание случая. В статье описывается клинический случай 33-летнего пациента, у которого отмечались рецидивирующие эпизоды сильной боли в животе, тошноты, рвоты, запора и онемения обеих нижних конечностей. Эти неспецифические нейровисцеральные приступы стали причиной ошибочных диагнозов острого аппендицита, синусовой тахикардии, камней в почках, острого лекарственного интерстициального нефрита и двух эпизодов частичной кишечной непроходимости. Шестой острый приступ вызвал подозрение на острую порфирию. Проба Уотсона-Шварца была положительной на порфобилиноген в моче. Анализ мутаций путем секвенирования ДНК, выделенной у пробанда, выявил ранее определенную миссенс-мутацию c.517C→ T, кодирующую p.R173W в гене HMBS , что подтверждает диагноз острой перемежающейся порфирии. Мутация также была выявлена у четырех из пяти членов семьи, которым был проведен таргетный мутационный анализ.
Заключение. Наиболее часто клиническая картина острой перемежающейся
порфирии характеризуется болью в животе с нейровисцеральными проявлениями, которые также наблюдаются при некоторых других соматических, психиатрических и хирургических патологиях. В связи с этим данное заболевание часто диагностируют неправильно, что приводит к неверному лечению и развитию тяжелых осложнений. Следовательно, для диагностики важны высокий уровень настороженности, а также осведомленность об основных лабораторных исследованиях. Точный диагноз
позволяет реализовать стратегии по предотвращению острых приступов, а также выполнить генетическое тестирование и провести консультирование членов семьи, относящихся к группе риска.
Сокращения
ОПП — острая перемежающаяся порфирия; ALA — аминолевулиновая кислота; ИМТ — индекс массы тела; ДНК — дезоксирибонуклеиновая кислота; HMBS — гидроксиметилбилансинтаза; PBG — порфобилиноген;
PBGD — порфобилиноген деаминаза; ПЦР — полимеразная цепная реакция.
Общие сведения
Острая перемежающаяся порфирия (ОПП) является острой порфирией с аутосомно-доминантным типом наследования. Заболевание вызвано
мутациями в гене, кодирующем гидроксиметилбилан-синтазу.
(HMBS) — третий фермент в пути биосинтеза гема. Название этого фермента используется как синоним порфобилиногендеаминазы (PBGD)[1]. На сегодняшний день выявлено более 400 мутаций в гене HMBS [2]. Наиболее частый клинический симптом — острая сильная боль в животе. При острых приступах у пациентов наблюдаются нейровисцеральные симптомы, включая рвоту, диарею, запоры, мышечную слабость, онемение, недержание или задержку мочи, сердцебиение, тремор и судороги, а также изменения поведения, такие как раздражительность, бессонница и эмоциональная лабильность.
Клинический случай
33-летний пациент поступил в специализированный госпиталь с перемежающимися болями в животе, тошнотой, рвотой, запором и онемением обеих нижних конечностей на протяжении 3-х дней. Он получал консервативное лечение в хирургическом отделении по поводу частичной кишечной непроходимости и ожидал диагностической лапароскопии. У пациента развилась спутанность сознания, также у него обнаружилась системная гипертензия, и его перевели в терапевтическое отделение для дальнейшего лечения. Пациент не страдает сахарным диабетом, артериальной гипертензии ранее не отмечалось. В анамнезе применение безрецептурных анальгетиков в течение 6 недель.
На протяжении 2-х последних лет были аналогичные нейровисцеральные приступы, потребовавшие пяти неотложных госпитализаций с постановкой различных клинических диагнозов. Шестой острый приступ вызвал подозрение на острую порфирию. Во время первого приступа в январе 2013 года был диагностирован аппендицит. Поскольку после операции симптомы ухудшились, было проведено экстренное лапароскопическое исследование.
Однако диагностическая лапароскопия не выявила причин, объясняющих
ухудшение симптомов. Второй приступ протекал в форме синусовой
тахикардии, и пациент начал получать бета-адреноблокаторы. Третий приступ, сопровождавшийся лихорадкой, был расценен как почечная колика и купирован консервативным лечением. Четвертый приступ был осложнен транзиторной гипонатриемией и преходящим повышением уровня креатинина в сыворотке. Врачи связывали эти осложнения с интерстициальным нефритом на основании того факта, что пациент принимал 50 мг диклофенака натрия два раза в сутки в течение 6 недель по назначению врача общей практики. Еще один приступ, произошедший в 2015 году, лечили как частичную кишечную непроходимость. Была выполнена диагностическая лапароскопия. Во всех этих случаях результаты ультразвукового исследования и диагностической лапароскопии не подтвердили хирургическую патологию. При осмотре пациент имел астеническое телосложение (ИМТ = 20 кг/м2), был бледным. Артериальное давление, измеренное на плечевой артерии, составило 160/90 мм рт. ст. На брюшной стенке имелись шрамы от предыдущих операций, в остальном без особенностей. Мышечная сила составила 4/5 во всех четырех конечностях (движение в полном объеме при действии силы тяжести и при небольшом внешнем противодействии).
отстаивании образец мочи постепенно становился темно-коричневым. Проба Уотсона-Шварца была положительной на порфобилиноген (PBG) в моче (рис. 1). Спектрофотометрия мочи на содержание общего уровня порфиринов показала «пик Соре». Общий уровень порфиринов в моче, рассчитанный с использованием скорректированной по Аллену абсорбции в образце мочи, составил 5505,5 нмоль/л (<300).

Генетические исследования проводились в зарубежной лаборатории. Полный анализ гена HMBS был выполнен с помощью ПЦР-амплификации выделенной ДНК с последующим экзон-специфическим анализом методом удлинения
праймеров для всех экзонов, границ экзонов/интронов и промоторных областей.
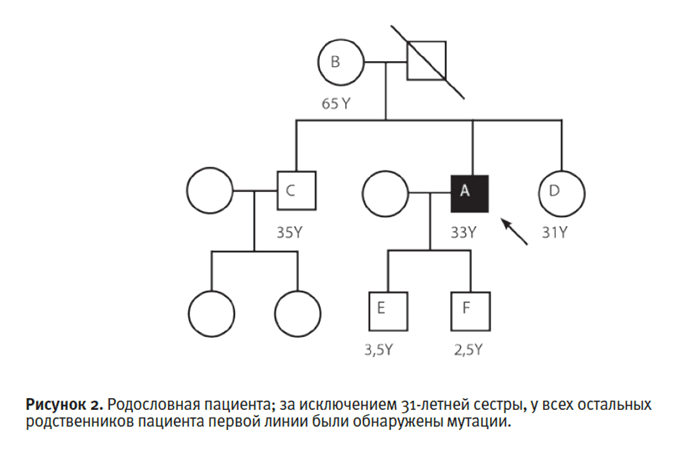
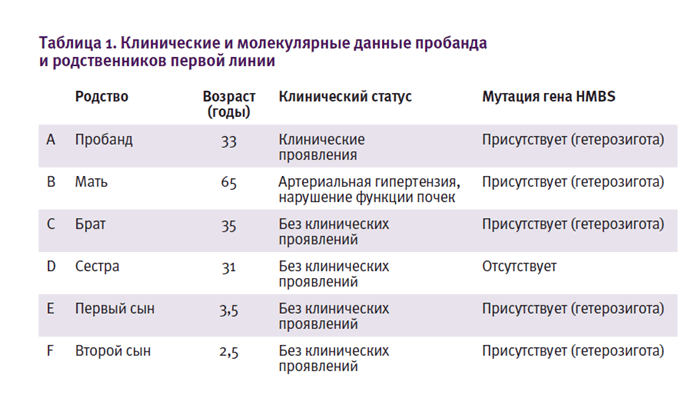
первой линии. Четыре из них были гетерозиготными по одной и той же
мутации гена HMBS (рис. 2 и табл.1).
Поскольку аргинат гема в Шри-Ланке недоступен, пациент получал только симптоматическое лечение. Углеводная нагрузка с внутривенным введением декстрозы и пероральным приемом углеводов была единственно возможным вариантом. Была проверена безопасность всех лекарственных препаратов, использованных для симптоматического лечения, на предмет риска острых
порфирий. Пациент был выписан из отделения, когда симптомы постепенно улучшились в течение 6 дней, для продолжения амбулаторного лечения. Оценить ответ на лечение не представлялось возможным из-за недоступности в Шри-Ланке количественных тестов для измерения аминолевулиновой кислоты (ALA) и PBG в моче.
Пациента проинформировали о факторах, провоцирующих развитие острой порфирии. Ему была предоставлена диагностическая карточка с информацией о лекарственных препаратах, применения которых следует избегать. Пациент
наблюдался в клинике с регулярным контролем функции почек, уровня гемоглобина и артериального давления. Наблюдение за пациентом в течение 1 года после постановки диагноза показало, что он перенес два легких приступа, которые не требовали стационарного лечения.
Исследования нервной проводимости не выполнялись, поскольку в интервалах между острыми приступами неврологических симптомов не наблюдалось. Родственникам, унаследовавшим мутацию HMBS, у которых не отмечалось клинических проявлений, также было рекомендовано избегать триггеров острых приступов, таких как определенные лекарственные препараты, голодание, алкоголь и гормоны. Брат пробанда получил консультацию относительно риска того, что его дети унаследуют мутацию HMBS, и рекомендацию выполнить генетическое исследование целевой мутации у обоих детей.
Несмотря на то что ОПП является наиболее распространенным типом
Поскольку порфирии встречаются редко, клиницисты нечасто сталкиваются с этими расстройствами, а учебная программа медицинских школ уделяет им очень мало внимания. Проба Уотсона-Шварца является скрининг-тестом для анализа мочи на PBG. 2 мл мочи смешивают с равным количеством реактива Эрлиха. Если присутствует PBG, он образует комплекс порфобилиноген-реактив Эрлиха — красный хромоген, который обычно нерастворим в хлороформе и N-бутаноле. При добавлении хлороформа PBG экстрагируется и придает красный цвет верхнему водному слою, тогда как комплекс уробилиноген-реактив Эрлиха экстрагируется в слой хлороформа. Поскольку у пациентов при острых приступах выделяются большие количества PBG, ложноотрицательные результаты во время острого приступа редки. Спектрофотометрия подкисленной мочи — это полуколичественный метод, который можно использовать в качестве скринингового теста для определения общего уровня порфиринов в моче. Все порфирины имеют полосу поглощения
при длине волны около 400 нм. Хотя в Шри-Ланке сложные методы не являются широкодоступными, простые надежные лабораторные тесты весьма эффективны для постановки диагноза порфирии [6], [7]. На сегодняшний день количественные тесты для определения уровней PBG, ALA, уропорфирина и копропорфирина, а также генетические исследования на порфирию в Шри-Ланке недоступны.
Окончательный диагноз порфирии следует устанавливать в зарубежной лаборатории. Молекулярно-диагностические исследования эффективны не только для биохимического подтверждения ОПП, но и для выявления членов семьи, подверженных риску [8]. По оценкам, пенетрантность острых приступов составляет примерно 1% у гетерозигот с вероятными патогенными вариантами HMBS [9]. Ранее сообщалось о мутации R173W в Швеции, России,
Испании, Великобритании, Японии и Китае. Мутация R173W по сравнению с другими распространенными мутациями может иметь более высокую клиническую пенетрантность и повышенный риск проявления заболевания. Эта миссенс-мутация представляет собой замену C-T в нуклеотиде 517 в экзоне 10. Превращение аргинина в триптофан в мутантном белке приводит к снижению активности фермента. Несмотря на то что активность PBGD в эритроцитах может быть использована в качестве показателя тяжести заболевания, у некоторых пациентов с острыми приступами его уровни могут быть нормальными [10]-[16]. Наиболее эффективным методом лечения острых
приступов является внутривенное введение аргината гема, который доступен в Европе и многих других странах мира, но не в Шри-Ланке.
Необходимо устранение провоцирующих факторов. При легких приступах может оказаться эффективной нагрузка углеводами. При проведении терапии необходимо контролировать уровни выделения ALA и PBG с мочой, чтобы оценить ответ. Поддерживающее лечение включает наркотические анальгетики, противорвотные средства, анксиолитики, восстановление и контроль баланса жидкости и психологическую поддержку. Стратегии борьбы с рецидивирующими острыми приступами подразумевают устранение потенциальных триггерных факторов и профилактические инфузии гемина [17, 18]. Однако данный пациент столкнулся с такими проблемами, как отсутствие аргината гема для неотложной терапии и профилактики, а также отсутствие контроля за ответом на лечение путем количественного измерения уровней ALA и PBG из-за недоступности и чрезмерно высокой стоимости этих средств. Поэтому длительное разрешение симптомов у этого пациента не представляется возможным. У пациентов с острой перемежающейся порфирией обычно отмечаются острые приступы с нейровисцеральными проявлениями, которые также наблюдаются при некоторых других соматических, психиатрических и хирургических патологиях. Таким образом, пациенты с порфирией, включая острые порфирии, потенциально могут столкнуться с проблемой недостаточной и неправильно диагностики, что значительно увеличивает время между появлением симптомов и подтвержденным диагнозом, приводит к неверному лечению и потенциально серьезным необратимым инвалидизирующим последствиям и даже вплоть до
Ошибочные клинические диагнозы у пациентов с острой порфирией могут стать причиной осложнений из-за возможного назначения порфирогенных препаратов [19]. При постановке неправильного хирургического диагноза проводятся ненужные хирургические вмешательства, такие как аппендэктомия и диагностическая лапароскопия.
поставлен психиатрический диагноз, могут возникнуть осложнения из-за некоторых антипсихотических средств, которые способны значительно ухудшить симптомы пациента с острой порфирией. Следовательно, для надлежащего установления диагноза важны высокий уровень настороженности, а также осведомленность о лабораторных исследованиях первой линии. Точный диагноз у пациента с ОПП позволяет реализовать стратегии по предотвращению острых приступов, а также провести скрининг и генетическое консультирование членов семьи, относящихся к группе риска.
Острая перемежающаяся порфирия
Острая перемежающаяся порфирия (ОПП) – тяжелое наследственное заболевание, характеризующееся нарушением синтеза порфиринов и накоплением их предшественников, оказывающих токсическое действие на различные органы и системы. Клинически проявляется сильной болью в животе, тошнотой, рвотой, тахикардией, гипертензией, полинейропатиями и психическими нарушениями. Диагноз ставится на основании определения повышенного содержания порфиринов и их предшественников в моче, снижения активности фермента порфобилиногендезаминазы в крови и ДНК-диагностики. Лечение заключается в подавлении образования порфиринов и симптоматической терапии.
МКБ-10
Общие сведения
Причины ОПП
В основе заболевания лежит генетически обусловленная неполноценность фермента порфобилиногендезаминазы (гидроксиметилбилансинтазы), участвующей в образовании порфиринов. Мутация расположена в гене PBGD на хромосоме 11 (локус 11q24.1-24.2). Порфирины – это большая группа органических соединений, которые являются составной частью белков, связывающих и переносящих кислород (гемоглобин, миоглобин), а также каталаз, расщепляющих перекись водорода, и цитохромов, обеспечивающих нейтрализацию ксенобиотиков (лекарства, алкоголь, яды). Синтез порфиринов осуществляется в костном мозге, печени, нейронах, эритроцитах. Дефицит порфобилиногендезаминазы при данном заболевании наблюдается главным образом в печени, а также в клетках нервной системы и эритроцитах. В результате недостаточной активности фермента происходит торможение образования порфиринов на уровне их предшественников - дельта-аминолевуленовой кислоты (ДАЛК) и порфобилиногена (ПБГ), которые являются токсичными для многих органов и систем.
Однако наличие неполноценного фермента не всегда приводит к развитию порфирии. Важную роль играют провоцирующие факторы, которые ускоряют выработку предшественников порфиринов. Такие факторы называются провоцирующими печеночными порфириногенами. Ими являются алкоголь, вирусные и бактериальные инфекции, стресс, хирургические операции, гипогликемия, голодание, колебания гормонального фона у женщин (менструация, беременность), воздействие тяжелых металлов (свинец, ртуть, висмут). Особое место отводится лекарственным препаратам, которые чаще всего вызывают манифестацию и обострение заболевания. Наиболее выраженным порфириногенным действием обладают препараты, активно метаболизирующиеся системой цитохрома P-450 - барбитураты, эстрогены, анальгин, парацетамол, гризеофульвин, сульфаниламиды, фенитоин, хлорамфеникол, карбамазепин, метилдопа.
Патогенез
В результате неполноценности фермента под действием провоцирующих факторов накапливаются промежуточные продукты метаболизма порфиринов, оказывающие свои повреждающие действия. Они блокируют натрий-калиевую АТФ-азу, тем самым изменяют метаболизм АТФ и медиаторов в нервных клетках. В результате этого развиваются демиелинизация и аксональная дегенерация нервных волокон. Эти патологические изменения больше выражены в периферической нервной системе, чем в центральной. Дистрофия в вегетативных ганглиях приводит к нарушению сосудистого тонуса и регуляции деятельности внутренних органов. Поражение абдоминальных сплетений вызывает спазм сосудов брыжейки и нарушение моторики желудка и кишечника. Вследствие ослабления активности блуждающего нерва усиливается влияние симпатической нервной системы на сердце и сосуды. Повреждение нервной системы также обусловлено возникающим дефицитом порфиринов в нейронах.
Предшественники порфиринов стимулируют выработку в гипоталамусе антидиуретического гормона. Возникает уменьшение диуреза, задержка жидкости, снижение осмолярности плазмы крови. Данное явление носит название синдром неадекватной секреции антидиуретического гормона (синдром Пархона - гидропексический синдром). Точный механизм развития этого синдрома при порфирии до сих пор неизвестен.
Классификация
В зависимости от наличия или отсутствия клинических проявлений, степени их выраженности, а также обнаружения специфических биохимических изменений в лабораторных исследованиях (повышение уровня общих порфиринов, ПБГ и ДАЛК в моче, снижение активности порфобилиногендезаминазы в эритроцитах) различают следующие формы острой перемежающейся порфирии:
Симптомы ОПП
Клинические проявления разнообразны. Чаще всего они возникают в молодом возрасте (около 20 лет). При латентной форме порфирии наблюдается следующая симптоматика – нарушение сна, склонность к артериальной гипертензии, учащенное сердцебиение, незначительные боли или неприятные ощущения в животе, снижение сухожильных рефлексов, мышечная слабость, психологические изменения личности.
При манифестной форме клиника развивается в виде острых атак. Продолжительность атаки составляет от 2 до 10 недель с рецидивами через 1-2 года. В начале приступа наиболее типичны проявления со стороны желудочно-кишечного тракта: интенсивные острые боли в животе, тошнота, рвота, вздутие, задержка стула. Боли имеют коликообразный характер, локализуются в разных отделах живота и бывают настолько сильными, что пациенты часто попадают в хирургический стационар с подозрением на острый аппендицит или перфорацию язвы желудка. Также повышается температура, увеличивается артериальное давление, вплоть до гипертонического криза, учащается сердцебиение. Характерным является розовая или красно-бурая окраска мочи.
Клиника поражения нервной системы присоединяется на 7-10 день приступа. Возникают разлитые боли по всему телу, снижается чувствительность на разных участках тела, выпадают сухожильные и кожные рефлексы. Появляются слабость и ограничение движения в конечностях (парез), более выраженные в проксимальных отделах – в плечах и бедрах. Иногда развивается полный паралич конечностей. В тяжелых случаях в патологический процесс вовлекаются черепно-мозговые нервы, что сопровождается двоением в глазах (диплопией), асимметрией лица, невнятной речью (дизартрией), нарушением глотания и поперхиванием (дисфагией).
Возможны судорожные припадки и психические нарушения – бессонница, эмоциональная лабильность, депрессивное состояние, неадекватное поведение, истерические припадки, зрительные и слуховые галлюцинации. Вследствие повышенной выработки антидиуретического гормона уменьшается мочеотделение, что приводит к водной интоксикации (гипоосмолярная гипергидратация), характеризующейся снижением аппетита, вялостью, адинамией, тремором, мышечными судорогами.
Осложнения
Наиболее тяжелые осложнения перемежающейся порфирии обусловлены полинейропатией. При параличе диафрагмы и межреберных мышц возникает острая дыхательная недостаточность, требующая проведения искусственной вентиляции легких. При слабости мышц глотки часть пищи может попасть в дыхательные пути и вызвать аспирационную пневмонию. В парализованных конечностях замедляется ток крови, что создает благоприятные условия для тромбообразования. Более редкие осложнения порфирии связаны с повышенным образованием антидиуретического гормона. Это отек головного мозга и рабдомиолиз (разрушение скелетных мышц). При рабдомиолизе из поврежденных мышечных клеток высвобождается миоглобин и калий, которые могут привести к острой почечной недостаточности и жизнеугрожающим нарушениям ритма сердца.
Диагностика
Пациентов с ОПП курирует врач-гематолог. Постановка диагноза перемежающейся порфирии представляет сложную задачу. Необходимо учитывать возраст пациента, время возникновения симптомов (например, лютеиновая фаза менструального цикла у женщин), наличие связи начала заболевания с провоцирующими факторами. Очень важно уточнять, какие лекарственные препараты принимал больной до развития приступа. Основная роль в распознавании перемежающейся порфирии отводится лабораторным методам:
Дифференциальный диагноз следует проводить с другими видами острых порфирий, демиелинизирующими заболеваниями (рассеянный склероз, синдром Гийена-Барре), отравлением свинцом, острыми абдоминальными хирургическими патологиями (аппендицит, кишечная непроходимость, холецистит, панкреатит, перфорация язвы желудка или двенадцатиперстной кишки), эпилепсией, психиатрическими болезнями. В дифференциальной диагностике принимают участие невролог, хирург, психиатр.
Лечение ОПП
Пациенты с манифестной и даже латентной формой подлежат лечению в гематологическом стационаре. При развитии выраженной неврологической симптоматики обязательна госпитализация в отделение реанимации и интенсивной терапии. Важно устранить все факторы, провоцирующие обострение заболевания. В первую очередь это касается приема лекарственных средств.
Этиотропной терапии не существует. Основная роль отводится патогенетическому лечению. Для этого применяются препараты, блокирующие образование токсичных предшественников порфиринов и, тем самым, уменьшающие их патологическое действие. К ним относятся большие дозы глюкозы, гема аргинат, сандостатин, аденил-5-монофосфат. Для ускорения регенерации миелиновой оболочки в нервных волокнах назначаются витамины группы В, для профилактики тромбообразования – антикоагулянты. Также используются антигипертензивные, анальгетические, противорвотные, слабительные, седативные препараты.
Если атаки порфирии являются менструалозависимыми и возникают часто (2-3 раза в год), необходимо подавление овуляции. С этой целью применяют агонисты гонадотропин-рилизинг гормона (Гозерелин). Беременность является неблагоприятным фактором и ассоциирована с молниеносным течением перемежающейся порфирии, высокой частотой летальных исходов. При развитии приступа в I и II триместре рекомендуется прерывание беременности, в III триместре проводится экстренное оперативное родоразрешение.
Прогноз и профилактика
Острая перемежающаяся порфирия - это тяжелое заболевание с неблагоприятным прогнозом и достаточно высоким уровнем летальности (15-20%). Самая частая причина смерти – паралич дыхательной мускулатуры вследствие полинейропатии. Очень важно своевременно диагностировать болезнь и начать специфическую терапию. Профилактика заключается в соблюдении высокоуглеводной диеты и избегании всех провоцирующих факторов, которые могут вызвать обострение – стрессов, инфекций, голодания, приема лекарственных средств и алкоголя. При наличии детей у пациентки с порфирией от новой беременности лучше отказаться. Всем родственникам больного порфирией для выявления скрытых или латентных форм заболевания необходимо проводить молекулярно-генетическую диагностику, определять уровень эритроцитарной порфобилиногендезаминазы и количество порфиринов в моче.
1. Диагностика и лечение острых порфирий. Клинические рекомендации национального гематологического сообщества/ под ред. Я.С. Пустовойт, С.К. Кравченко, Р.Г. Шмакова, В.Г. Савченко. - 2018.
2. Лабораторная диагностика острой перемежающейся порфирии/ Карпова И.В., Сурин В.Л., Тагиев А.Ф., Пивник А.В.// Проблемы гематологии и переливания крови. - 1998 - №1.
3. Острая порфирия с полиневропатией и положительным эффектом лечения глюкозой/ Котов С.В., Сидорова О.П.// Медицинская генетика. – 2016 - №7.
4. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена: Моногр./Б.Н.Кривошеев и др.
Читайте также: