Геномный импринтинг как причина рака
Добавил пользователь Morpheus Обновлено: 20.01.2026
■ Геномный импринтинг —эпигенетический механизм регуляции экспрессии гомологичных генов развивающегося организма в зависимости от их родительского происхождения. В статье рассмотрены патологические состояния, наследственные болезни и синдромы, обусловленные нарушением работы импринтированных генов вследствие их однородительского наследования, гемизиготного состояния, а также аномалий эпигенетического маркирования.
В геноме человека отцовские и материнские гены могут обнаруживать дифференциальную активность уже на ранних стадиях онтогенеза. При этом наблюдается видимое искажение менделевских правил наследования отдельных признаков. В участках генома, подверженных импринтингу (от англ. imprint — отпечаток, запечатление), экспрессируется только одна аллель (аллель — альтернативное состояние гена) — отцовская или материнская. Иными словами, экспрессия импринтированного гена в организме-потомке определяется его родительским происхождением, то есть зависит от того, передается ли он геномом спермия или яйцеклетки.
При болезнях генного импринтинга наблюдается моноаллельная экспрессия в локусах хромосом одного из родителей. Причина - точковые мутации в генах, дифференцированно экспрессирующихся в зависимости от материнского и отцовского происхождения и приводящих к специфическому метилированию цитозиновых оснований в молекуле ДНК.
Эти мутации обусловливают развитие заболеваний, для которых большое значение имеют характер наследования и происхождение хромосом. К таким заболеваниям относятся:
• болезнь Гиршпрунга, обусловленная мутацией в гене RET (10q11.2); чаще всего наследуется по материнской линии;
• нейрофиброматоз Реклингаузена (тип 2) - мутация в гене SCH (22q12); наследуется по материнской линии;
• псориаз - проявляется тяжелее, если наследуется по отцовской линии;
• семейная гипертрофическая кардиомиопатия - наследуется по материнской линии;
• синдром Ангельмана (СА) - делеция критического района, находящегося в материнской хромосоме 15 (15q11.2-q13);
• синдром Вильямса - проявляется более выраженной задержкой физического и умственного развития и микроцефалией, если делеция затрагивает материнскую хромосому 7 (7q11.23);
• синдром «крика кошки» - проявляется более выраженно, если делеция захватывает отцовскую хромосому 5 (5р15.3);
• синдром Корнелии де Ланге (3q26) - проявляется более выраженно, если наследуется по материнской линии;
• синдром Прадера-Вилли (СПВ) - делеция критического района, находящегося в отцовской хромосоме 15 (15q11.2-q13);
• синдром Турета и поликистоз почек - проявляются раньше и тяжелее, если наследуются по материнской линии;
• тяжелая (злокачественная) шизофрения - проявляется более выраженно, если наследуется по отцовской линии;
• spina bifida - наследуется по материнской линии (в 2 раза чаще, чем по отцовской линии);
• эпилепсия - проявляется тяжелее, если наследуется по материнской линии.
Следует отметить, что в случае СА и СПВ особенности молекулярной организации критического района хромосомы 15 связаны с гомологической рекомбинацией, мейотической нестабильностью и объясняют высокую частоту хромосомных аберраций. В свою очередь, в семьях с повторными случаями СА и СПВ в геномах здоровых родственников обнаружены близко расположенные, но противоположно импринтированные гены, которые названы генамикандидатами этих заболеваний (см. ниже).
Кроме того, обнаружена потеря фрагментов хромосом в клетках злокачественных опухолей, расцениваемая как потеря гетерозиготности (см. главы 17 и 25). Например, при нейробластомах утрачивался локус 1р36 материнского происхождения или наблюдалась гиперамплификация гена N-Myc в локусе 2р24 отцовского происхождения.
Механизмы патогенеза
Известны два механизма патогенеза, касающиеся как самой молекулы ДНК, так и молекул белков, входящих в состав хроматина хромосом, подверженных импринтингу.
Первый механизм - это нарушение метилирования импринтированных генов. В настоящее время хорошо изучена эпигенетическая модификация или специфическое метилирование цитозиновых остатков ДНК по 5-му углеродному атому. Это единственная допустимая в физиологических условиях химическая модификация, стабильно сохраняющаяся в ряду клеточных поколений и прямо или косвенно влияющая на экспрессию генов. У человека дифференцированное метилирование родительских аллелей наблюдается, как правило, внутри или рядом с ГЦ-богатыми районами, содержащими разные типы нуклеотидных повторов, между которыми нет гомологии, а длина единицы повтора каждый раз другая, и возможно его любое расположение по отношению к импринтированному гену, промотору или регуляторному участку. По-видимому, такие повторы вовлекаются в установку процесса импринтинга (метилирования гена). Они
служат мишенями для маркирования определенного аллеля за счет организации уникальной для него вторичной структуры ДНК. Так, показано, что эти повторы создают свернутые структуры, узнаваемые гетерохроматинспецифическими белками. Например, метилирование CpG-районов изменяет структуру ДНК с образованием формы z-ДНК, что ведет к полной инактивации импринтированных генов.
Второй механизм связан с особенностями структурной организации и функционирования хроматина в локусах, в которых располагаются импринтированные гены. В пользу этого указывают результаты экспериментов по изучению времени репликации импринтированных хромосомных доменов в S-фазе митоза. В частности, до репликации в клеточном ядре наблюдаются два гибридизационных сигнала, соответствующих материнскому и отцовскому аллелям импринтированного гена, а после репликации такой сигнал приобретает сдвоенную структуру. Асинхронность вычисляется как соотношение сдвоенных и одиночных сигналов.
В случае СПВ и СА критический район соответственно на отцовской или материнской хромосоме 15 раньше реплицируется в S-фазе. При этом время репликации коррелирует с уровнем активности генов и зависит от конденсации хроматина в районах промоторов и примыкающих к нему районах.
Инактивация транскрипции сопровождается уплотнением хроматина (гетерохроматизацией), в результате чего ДНК становится менее доступной для РНК-полимеразы и факторов, необходимых для инициации транскрипции. Химическая природа модификаций гетерохроматина до сих пор не выяснена. Имеются данные о взаимосвязи процессов упаковки хроматина и метилирования ДНК. Например, показано, что транскрипционно активный хроматин имеет пониженное содержание линкерного гистона Н1, связывающего между собой нуклеосомы и упаковывающего их в фибриллы (см. главу 3). Этот гистоновый белок предпочтительно связывается с метилированными последовательностями ДНК.
Такое же сродство (аффинность) имеют негистоновые белки хроматина группы МеСР. При этом сила связывания определяется плотностью метилированных CG-динуклеотидов, а не конкретной нуклеотидной последовательностью. Некоторые из негистоновых белков хроматина подавляют транскрипцию непосредственно, например в белке МеСР2 для этого имеется специальный домен.
Таким образом, установка процессов метилирования ДНК про-
исходит только в импринтированных локусах на последующих этапах дифференцировки гамет. Основным ферментом, обеспечивающим метилирование de novo у млекопитающих, является ДНКметилтрансфераза, или Dnmt1 (см. главу 7). Вместе с тем, остается невыясненным механизм распознавания нуклеотидных последовательностей ДНК, которые должны быть по-разному метилированы в отцовском и материнском гаметогенезе. В этой связи были выделены два альтернативных способа сплайсинга 5'-экзонов гена Dnmt1, один из которых реализуется в оогенезе, другой - в сперматогенезе.
При прямом эпигеномном воздействии на экспрессию конкретного гена метилированию подвергается сам импринтированный ген. В этом процессе участвуют ДНК-связывающие белки, вызывающие гетерохроматизацию метилированного локуса. В результате доступ активаторов транскрипции к ДНК ограничивается, и экспрессия гена останавливается. При этом действие многочисленных факторов транскрипции зависит от характера метилирования ДНК. Среди этих факторов выделяют, с одной стороны, метилчувствительные активаторы и метилзависимые репрессоры (они опосредуют инактивацию метилированного гена), а с другой стороны - метилчувствительные репрессоры и метилзависимые активаторы (они обеспечивают экспрессию метилированного гена).
В случае косвенного влияния метилирования на экспрессию импринтированного гена предполагается модификация не самого гена, а другого гена - импринтора, находящегося на той же хромосоме в цис-положении. При этом функция гена-импринтора направлена на поддержание моноаллельной экспрессии одного или нескольких импринтированных локусов в пределах конкретного кластера генов.
Как оказалось, гены-импринторы часто (если не всегда) кодируют нетранслируемые РНК - это универсальный механизм конкурентной экспрессии, необходимый для поддержания в соматических клетках моноаллельной экспрессии всех импринтированных генов, включая гены, относящиеся к кластеру генов СПВ и СА.
Таким образом, общей особенностью импринтированных генов, находящихся в составе критических районов хромосом, является наличие генов, кодирующих нетранслируемую РНК. Например, для СПВ и СА обнаружено несколько таких генов (ZNF127AS, PAR5, PARSN, IPW, PAR1, C15orf2, PWCR1, UBE3A-AS). Некоторые из них синтезируются на антисмысловой цепи соответствующих генов (AS
означает «антисенс» - см. главу 20). В частности, на антисмысловой цепи белок-кодирующего гена ZNF127 транскрибируется в противоположном направлении нетранслируемая антисмысловая РНК, или ZNF127AS. Причем ген ZNF127 и его антисмысловой аналог ZNF127AS активны только на отцовской хромосоме.
Антисмысловая РНК также обнаружена для гена UBE3A, но его транскрипция (как и UBE3A-AS) происходит на разных родительских хромосомах. Так, UBE3A-AS экспрессируется на отцовской хромосоме и только в тех тканях мозга, в которых UBE3A подвержен импринтингу и активен только на материнской хромосоме. В остальных тканях, где UBE3A экспрессируется биаллельно, транскрипт UBE3A-AS не обнаруживается.
В целом можно заключить, что механизмы генного импринтинга остаются малоизученными.
Геномный импринтинг как причина рака
Инверсии и транслокации генов как причина рака
Инверсии генов при раке. Возможно, инверсии представляют собой наименее распространенный вид мутаций. При данном виде нарушений участок ДНК образует петлю и меняет свое направление: 3'-конец становится 5'-концом. Размер инвертированных участков, разумеется, варьирует.
Наиболее важная инверсия из тех, что вызывают заболевания у человека, происходит в гене фактора VIII. Она ведет к развитию тяжелой гемофилии А. На сегодня злокачественные опухоли, причиной которых могли бы быть инверсии, не обнаружены.
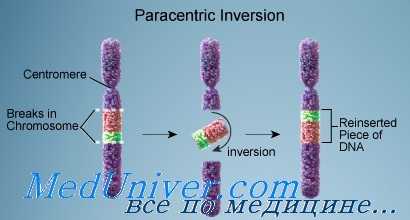
Парацентрическая инверсия
Транслокации генов при раке
В клетках злокачественных опухолей описано более 100 часто встречающихся транслокаций. Многие из них постоянно выявляются при определенных опухолях, что свидетельствует об их вовлечении в процесс злокачественной трансформации.
Некоторые транслокации могут быть вторичными в развитии более агрессивных фенотипических изменений. Генетическая нестабильность, присущая злокачественным клеткам, ведет к дальнейшему усилению кариотипических аномалий в процессе прогрессирования болезни, а дополнительные генетические нарушения повышают пролиферативный потенциал опухоли.
Единственная транслокация, как правило, не вызывает немедленное развитие опухоли; это показано на примере больных атаксией-телеангиэктазией, имеющих высокий риск лейкоза. У этих больных за много лет до развития злокачественного процесса можно выявить лимфоциты с типичной транслокацией. В таблице приведены некоторые примеры транслокаций при солидных опухолях.
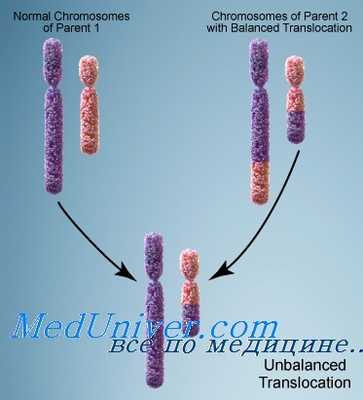
Схема транслокации
Генетическая нестабильность — распространенное явление при злокачественных новообразованиях. Наиболее ярко это выражено при ННПКРР, известном также как синдром Линча типа II.
Заболевание характеризуется нестабильностью микросателлитных последовательностей и связано с дефектами нескольких генов, ответственных за репарацию некомплементарных спаривании нуклеотидов. Частота мутаций в клетках с нарушениями таких генов значительно выше по сравнению с нормальными генами в интактных клетках.
Опухоли больных ННПКРР могут служить моделью для изучения спорадических онкологических заболеваний, т. к. во всех этих случаях развиваются сходные мутации, поражающие одни и те же гены и последовательности ДНК. Так, Krawczak показал, что спектр нуклеотидных замен в гене ТР53 при спорадических новообразованиях весьма близок к нарушениям, определяемым в опухолях больных с наследственными онкологическими синдромами.

Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Причиной психических заболеваний может быть конфликт между отцовскими и материнскими генами
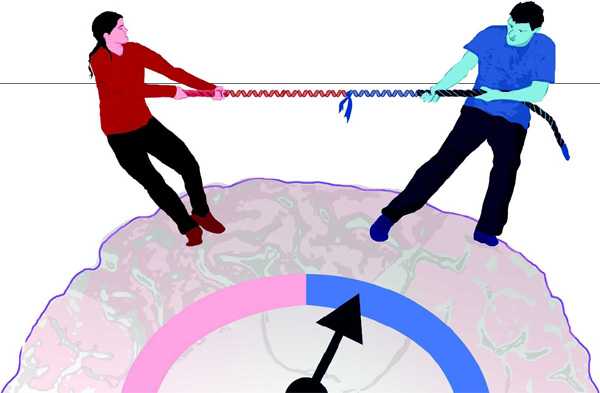
Согласно новой гипотезе, причины многих психических заболеваний кроются в своеобразном «перетягивании каната», в которое вовлечены в человеческом организме гены, унаследованные от отца и матери. Рис. из обсуждаемой статьи в Nature
В основе многих психических заболеваний, включая аутизм, паранойю и депрессию, может лежать конфликт между генами, унаследованными от отца и матери. Известно, что самцы млекопитающих «отключают» в своих сперматозоидах многие гены, активная работа которых выгодна матери, но невыгодна эмбриону, тогда как самки отключают в своих яйцеклетках гены с противоположным эффектом. Ряд фактов свидетельствует о том, что смещение баланса активности генов в «отцовскую» сторону может приводить к аутизму, в «материнскую» — к паранойе, депрессии и психозам.
Журнал Nature опубликовал краткое эссе британского социолога Кристофера Бэдкока (Christopher Badcock) и канадского биолога Бернарда Креспи (Bernard Crespi), в котором авторы предприняли смелую попытку дать единое объяснение широкому спектру психических отклонений, включая аутизм, паранойю, депрессию, шизофрению и различные психозы. Давно известно, что эти заболевания имеют в значительной степени наследственную природу, однако закономерности их наследования не всегда укладываются в стандартные генетические модели. Бэдкок и Креспи предположили, что причины этих отклонений кроются не столько в самих генах, которые ребенок получает от отца и матери, сколько в балансе активности (экспрессии) этих генов.
У млекопитающих, включая человека, широко распространен так называемый геномный импринтинг. Суть явления в том, что некоторые гены в половых клетках родителей особым образом «метятся» (например, путем метилирования цитозиновых оснований). «Помеченный» ген у потомства просто-напросто не работает. Некоторые гены отключаются в сперматозоидах, другие в яйцеклетках. В результате часть признаков потомство наследует только от матери (если соответствующие гены отключены в сперматозоидах), часть — только от отца (если ген отключен в яйцеклетке). В половых клетках потомства старые метки могут быть удалены и заменены новыми. В результате у внуков могут проявиться признаки деда или бабки, которые не были выражены у родителей. Импринтинг — это пример так называемой «эпигенетической», или надгенетической, наследственности, то есть наследственных свойств, не связанных с изменением основной структуры генов — последовательности нуклеотидов в молекулах ДНК.
Предполагается, что геномный импринтинг развился из-за конфликта интересов между полами. У млекопитающих между самкой и ее детенышем во время внутриутробного развития складываются отчасти антагонистические отношения. Говоря упрощенно, эмбрион старается высосать из матери побольше соков, а мать старается сохранить силы и здоровье, чтобы в будущем иметь возможность родить и других детей. Самец в этом конфликте в общем случае — на стороне детеныша. Других детей самка еще неизвестно от кого родит, а этот — свой. Поэтому самцы отключают в своих сперматозоидах те гены, которые защищают мать от чрезмерных притязаний эмбриона, а матери, напротив, отключают в своих яйцеклетках те гены, которые могут эти притязания усилить. Действительно, большинство генов, подвергающихся родительскому импринтингу, так или иначе связаны с внутриутробным развитием, строением плаценты и т. п.
Например, человеческий зародыш получает и от отца, и от матери по одной копии гена IGF2, который кодирует белок, способствующий быстрому росту. В норме отцовская копия этого гена активна, а материнская отключена. В данном случае, как и во многих других, геномный импринтинг работает в точном соответствии с теорией. Мать, отключая в своих яйцеклетках ген IGF2, старается притормозить рост ребенка и тем самым облегчить себе жизнь во время беременности, родов и грудного вскармливания. Отец, снабжая ребенка активной копией гена, напротив, старается ускорить рост эмбриона вопреки «корыстным» интересам матери.
Таким образом, материнские и отцовские гены в организме зародыша играют в игру, напоминающую перетягивание каната. В результате этой игры в ходе эволюции устанавливается некий баланс, который и обеспечивает нормальное развитие эмбриона (например, отключение гена одним из родителей может быть уравновешено усилением активности его работающей копии в результате мутаций и отбора, и т. п.). Нарушение этого баланса чревато неприятными последствиями. Так, если у человеческого зародыша активными оказываются обе копии гена IGF2 (то есть происходит сдвиг баланса активности генов в «отцовскую» сторону), ребенок будет страдать так называемым синдромом Беквита–Видемана (Beckwith–Wiedemann syndrome), для которого характерен резко увеличенный вес ребенка при рождении и чрезмерно быстрый рост некоторых органов. Дети, у которых баланс смещен в «материнскую» сторону (отключены обе копии гена IGF2), страдают синдромом Сильвера–Рассела (Silver–Russell syndrome), характерным признаком которого является замедленный рост.
Считается, что несколько сотен из примерно 20000 человеческих генов подвергаются геномному импринтингу, хотя точно это установлено пока лишь для 63 генов. Большинство из них оказывают серьезное влияние на рост и развитие ребенка. Особенно часто подвергаются импринтингу гены, от которых зависит работа плаценты — органа, отвечающего за изъятие эмбрионом ресурсов из материнского организма. Кроме того, импринтингу подвергается ряд генов, связанных с развитием мозга. Это значит, что генетическое «перетягивание каната» в принципе может влиять на поведение, мышление и свойства личности.
Авторы эссе упоминают идеи крупнейшего эволюциониста-теоретика XX века Уильяма Гамильтона (William Donald Hamilton), автора концепции «эгоистичного гена». Эта концепция получила всемирную известность благодаря работам Ричарда Докинза (см.: Р. Докинз. Эгоистичный ген). Именно Гамильтон первым предположил, что важнейшей движущей силой эволюции может быть конфликт между генами, в том числе и между генами одного и того же организма. Гамильтон также допускал, что внутренний генетический конфликт может иметь психологические последствия. Он отмечал, что людей можно подразделить на две группы: «people–people» и «things–people», то есть на людей с мышлением, ориентированным на других людей, и людей с мышлением, ориентированным на вещи. Сам себя он относил ко второй группе и полагал, что хотя такой психический склад чреват асоциальностью, именно он позволил ему добиться успеха в науке. По мнению авторов эссе, Гамильтон страдал легкой формой аутизма.
Авторы полагают, что ярко выраженная склонность к «вещевому» мышлению — это не что иное, как аутизм. Аутичные дети в раннем возрасте отличаются замкнутостью на себе и крайним эгоизмом и доставляют очень много хлопот своим воспитателям (в первую очередь, конечно, матери). Это навело авторов на мысль, что некоторые случаи аутизма могут объясняться сдвигом в «отцовскую» сторону баланса активности генов, связанных с развитием мозга. Это предположение в дальнейшем получило фактическое подтверждение. Оказалось, что у людей с синдромом Беквита–Видемана риск развития аутизма резко повышен. Кроме того, для многих аутистов, как выяснилось, характерна повышенная активность гена IGF2. Эти данные показывают, что между аутизмом и геномным импринтингом действительно может существовать какая-то связь.
Если сдвиг баланса активности генов в «отцовскую» сторону ведет к аутизму, то его сдвиг в «материнскую» сторону, по идее, должен давать некий противоположный эффект. Какое же психическое состояние можно считать противоположным аутизму? По мнению, авторов, подлинным «антиподом» аутизма является паранойя, а также ряд других психических отклонений со сходными симптомами. Например, аутисты обычно не замечают или не обращают внимания на то, что за ними кто-то наблюдает; параноики, напротив, болезненно чувствительны к подобным вещам, им часто мерещится, что за ними наблюдают, следят, шпионят. Аутисты не способны к координированным действиям в группе, они не способны понять причины, мотивы и механизмы коллективной деятельности. Параноики, напротив, болезненно сосредоточены на подобных действиях — реальных или воображаемых, для них характерны бредовые идеи конспирологического характера, в любом событии они склонны видеть происки каких-то тайных группировок. В целом для аутистов характерно слабое развитие так называемой «теории разума» (они не могут понять мотивы, мысли и желания других людей), тогда как у параноиков, наоборот, «теория разума» в определенном смысле гипертрофирована. Именно поэтому параноики усматривают разумный замысел, чье-то сознательное вмешательство или сокровенный смысл чуть ли не во всём, что их окружает (этим обусловлены их многочисленные религиозные, магические и мистические иллюзии). Многие другие симптомы аутизма и паранойи, по мнению авторов, тоже представляют собой диаметральные противоположности.
Авторы предполагают, что симптомы аутизма в общем и целом проистекают из недостаточного развития «теории разума» (это состояние они называют «гипоментализмом»), тогда как для паранойи и широкого круга психотических отклонений характерно противоположное состояние — «гиперментализм».
По мнению авторов, небольшие отклонения баланса геномного импринтинга в «материнскую» сторону должны вести не только к замедлению роста детей, что делает их вынашивание и вскармливание более «дешевым», но и к таким изменениям в поведении и мышлении, которые облегчают матери заботу о них. Такие дети должны быть более чуткими, менее требовательными, у них должна быть сильнее развита способность чувствовать психическое состояние близких и тонко реагировать на него. Сильное смещение баланса в «материнскую» сторону приводит к психозу. Напротив, небольшое смещение баланса в «отцовскую» сторону должно менять поведение детей в сторону большей требовательности, эгоизма, сосредоточенности «на вещах». Сильное смещение в «отцовскую» сторону ведет к тяжелому «гипоментализму» и аутизму. Посередине между этими крайностями лежит вожделенная «норма».
Исследователи приводят в подтверждение своей гипотезы ряд фактов. Так, генетики обнаружили на 15-й хромосоме человека участок, содержащий несколько генов, подвергающихся импринтингу. Дети, у которых баланс активности этих генов сдвинут в «отцовскую» сторону, страдают синдромом Ангельмана. Такие дети в раннем возрасте отличаются гиперактивностью и постоянно требуют внимания к себе. Среди них очень высок процент аутистов. Дети, у которых баланс сдвинут в «материнскую» сторону, страдают синдромом Прадера–Вилли. Они отличаются сонливостью, исключительным спокойствием и нетребовательностью в раннем возрасте; в дальнейшем большинство из них начинает страдать различными психозами и депрессиями.
По мнению авторов, существует два основных фактора, от которых зависит соотношение гипо- и гиперменталистских тенденций в человеческом мышлении. Первый фактор, как мы уже знаем, это активность генов, подвергающихся родительскому импринтингу. Второй фактор — пол. Для мужчин в целом более характерен гипоментализм и «ориентированность на вещи», для женщин — гиперментализм и «ориентированность на людей».
Между прочим, недавно была показана сильная положительная корреляция между концентрацией мужского полового гормона тестостерона в крови эмбриона во время внутриутробного развития и склонностью ребенка к аутизму. Иными словами, авторы полагают, что мышление мужчин в норме чуть-чуть склоняется в сторону аутизма, а женщин — в сторону паранойи. Это согласуется с тем, что аутизм чаще встречается у мужчин, чем у женщин. Авторы также замечают, что их теория помогает объяснить и другой факт, до сих пор остававшийся необъясненным: хотя женщины страдают аутизмом реже мужчин, заболевание у них обычно протекает в более тяжелой форме. Напротив, шизофрения и депрессия у мужчин обычно протекают тяжелее, чем у женщин.
Возможно, считают авторы, психическое заболевание развивается чаще, но протекает в более легкой форме, когда направление действия двух факторов — половой принадлежности и баланса импринтинга — совпадают, как у мужчин в случае аутизма или у женщин при депрессии. Когда же сбой геномного импринтинга направляет психическое развитие человека в сторону, противоположную той, к которой он предрасположен в силу своей половой принадлежности, заболевание развивается реже, но протекает в более тяжелой форме, как у женщин-аутистов и мужчин, страдающих психозами.
Для проверки гипотезы необходимо детальное изучение всех человеческих генов, подвергающихся родительскому импринтингу, и выявление функции этих генов. Эта работа сейчас продвигается весьма успешно, а методы исследований быстро совершенствуются. Если интересная и смелая гипотеза о единой природе широкого спектра психических заболеваний подтвердится, можно будет подумать даже о разработке универсальных методов их лечения и профилактики. Ведь в фармакологической корректировке активности генов нет ничего невозможного — технически это гораздо проще, чем изменить нуклеотидную последовательность какого-нибудь гена во всех клетках организма.
Источник: Christopher Badcock, Bernard Crespi. Battle of the sexes may set the brain // Nature. 2008. V. 454. P. 1054–1055.
Доказано, что ряд вирусов принимает участие в патогенезе злокачественных новообразований. Около 30 лет назад выявили четкую связь между инфицированием вирусом Эпштейна-Барр и риском лимфомы Беркитта и рака носоглотки. Вместе с тем изолированное поражение данным вирусом недостаточно фактором для развития указанных новообразований.
В меньшей степени вирус Эпштейна-Барр способствует развитию смешанно-клеточного варианта лимфогранулематоза, лимфоидного истощения и Т-клеточных лимфом. Последнее обстоятельство особенно интересно, поскольку вирус Эпштейна-Барр обычно не поражает Т-лимфоциты. Таким образом, данный вирус способствует развитию В-клеточных лимфопролиферативных заболеваний у лиц с иммунодефицитом, особенно со СПИДом.
Другим примером роли вирусов в онкогенезе служит 200-кратное повышение риска печеночно-клеточного рака у взрослых и детей с хроническим вирусным гепатитом В. Латентный период от момента инфицирования вирусом до развития печеночноклеточного рака у взрослых составляет около 20 лет, однако в случае внутриутробного заражения плода первые признаки новообразования способны развиться через 6-7 лет. Дополнительные факторы онкогенеза в данном случае окончательно не выяснены. Риск печеночноклеточного рака и лимфомы селезенки повышен также при инфицировании вирусом гепатита С.
Вирус папилломы человека обнаруживают у большинства больных раком шейки матки. Особенно опасны в этом отношении вирусы папилломы типов 16, 18 и (реже) 31, 33, 35, 45 и 56. Вместе с тем вирусы папилломы типов 6 и 11, вызывающие остроконечные кондиломы, имеют крайне низкий онкогенный потенциал. По аналогии с другими сходными заболеваниями инфицирование вирусом не является достаточным фактором для развития злокачественного новообразования.
Механизм онкогенного действия вируса папилломы человека типа 19 связан с мутацией генов — супрессоров опухолевого роста (Р53 и RB) и последующим нарушением нормального цикла деления клетки на границе периодов G, и S, а также на границе периода G2 и митоза.
Герпесвирус человека типа 8 повышает вероятность развития саркомы Капоши, первичной лимфомы серозных оболочек, плазмоклеточного варианта синдрома Каслмана, которые наиболее распространены у лиц со СПИДом. Т-лимфотропный вирус человека типа 1 сочетается с Т-клеточным лейкозом/лимфомой взрослых.
Геномный импринтинг
В ряде случаев онкогенез связан с геномным импринтингом — избирательной инактивацией одного или двух аллелей определенного гена. Инактивированный ген может наследоваться как от отца, так и от матери. К примеру, в норме материнский ген ИФР-2 (рецептор к инсулиноподобному фактору роста типа 2) инактивирован в результате метилирования группы цитозин-гуанин (Ц-Г), расположенной неподалеку от промотора данного гена.
Транскрипция гена в подобных условиях невозможна. В ряде случаев у больных с опухолью Вильмса наблюдают потерю метилирования материнского гена ИФР-2, что, в свою очередь, проявляется его нормальной транскрипцией. В то же время ген Н19, функция которого до конца не изучена, приобретает указанную метильную группу, что приводит к торможению его транскрипции. Синдром Беквита-Видемана, который проявляется макросомией, макроглоссией, гемигипертрофией, грыжей пупочного канатика и аномалией почек, сочетается с повышенным риском опухоли Вильмса, гепатобластомы, рабдомиосаркомы, нейробластомы и рака коры надпочечников. Повышенный риск развития рака связан также с нарушением метилирования генов в локусе lip 15.
Теломераза
Теломера — это концевые участки хромосомы, которые выполняют функцию ее стабилизации, защиты от повреждений и транслокаций. Теломеразная последовательность ДНК состоит из нескольких десятков или тысяч повторов шести нуклеотидов — TTAGGG. В процессе репликации ДНК происходит прогрессирующее укорочение длины теломеры — проявление старения клетки. Экспрессия теломеразы — обратной транскриптазы, обеспечивающей репликацию теломер, — приводит к значительному увеличению числа потенциальных делений клетки.
Терапия, направленная на торможение теломеразы, приводит к гибели опухолевых клеток.
Генетика и онкология: главные вопросы
Что такое онкоген? Как возникают мутации в ДНК? Какие мутации провоцируют рак? Кому и чем могут помочь молекулярно-генетические исследования?
На эти и другие вопросы во Всемирный день ДНК отвечает Александр Олегович Иванцов – доктор медицинских наук, старший научный сотрудник научной лаборатории морфологии опухолей ФГБУ «НМИЦ онкологии им. Н.Н. Петрова» Минздрава России.

Александр Олегович Иванцов, доктор медицинских наук
— Александр Олегович, что такое мутация? Как возникают «поломки» в молекулах ДНК?
— Организм человека состоит из большого числа специализированных клеток, ядра которых содержат нуклеиновые кислоты: ДНК и РНК. Совокупность этих молекул содержит биологическую информацию, необходимую для построения и поддержания клеток, органов и систем органов в целом. Весь наследственный материал, заключённый в клетке, получил название – геном. У человека он представлен 23 парами хромосом (22 пары аутосом и пара половых хромосом), находящихся в ядре. ДНК является длинной полимерной молекулой, она хранит биологическую информацию в виде генетического кода, состоящего из последовательности повторяющихся блоков — нуклеотидов. Последовательность нуклеотидов позволяет «кодировать» информацию о различных типах РНК, которые необходимы для последующего биосинтеза важнейших белков. Открытие структуры ДНК в 1953 году стало поворотным моментом в развитии биологии, а исследователям Фрэнсису Крику, Джеймсу Уотсону и Морису Уилкинсу была присуждена Нобелевская премия в 1962 году. Стойкое изменение генома получило название – мутация. Эти изменения могут касаться структуры отдельных генов, хромосом и генома в целом. То есть изменение последовательности нуклеотидов приводит к нарушениям в кодировании информации – в итоге к аномалиям на уровне качества или количества соответствующих белков.
— Почему некоторые мутации приводят к развитию рака? Как устроен этот механизм? Как ученые определяют, какие именно «поломки» в ДНК приводят к развитию злокачественных опухолей?
— Чтобы ответить на этот вопрос, стоит разобраться как развивается опухоль. Она имеет автономный характер роста. Что это значит? В норме количество клеток в организме человека регулируется балансировкой двух противоположных процессов – клеточного деления и клеточной гибели. При росте опухоли прибавление клеточной массы опережает клеточную гибель. Это возможно по двум причинам – либо активируются процессы пролиферации, т.е. деления клетки, либо угнетается апоптоз, т.е. запрограммированная клеточная гибель. Автономность опухоли состоит в том, что ее клетки не способны реагировать на внешние сигналы организма, и, как следствие, она продолжает рост.
Если изменения нуклеотидной последовательности ДНК происходят в значащих фрагментах ДНК (прим. – экзонах), то они могут привести к развитию опухоли. К развитию рака приводят в основном мутации, нарушающие баланс деления и гибели клеток, то есть мутации в генах, контролирующих именно эти процессы. Мутации могут возникать случайно, например, в процессе удвоения ДНК в результате деления клетки. А могут возникать под влиянием мутагенов: например, воздействия ультрафиолетового или рентгеновского излучения, высокой температуры, некоторых химических веществ. На последний вопрос, можно ответить, что патогенность мутации можно предположить в первую очередь по функции гена, который она затрагивает, по её структурным характеристикам (насколько сильно она нарушает или изменяет работу этого гена), и подтвердить путем функциональных исследований (например, на клеточных культурах).
— Что такое онкогены?
— Онкогеном называется ген, который в норме не оказывает влияние на процессы деления и гибели клеток, а в опухоли активизируется, вследствие чего раковые клетки приобретают способность к неконтролируемому размножению. Кроме того, в настоящее время известно о роли антионкогенов. В норме они подавляют процесс деления клеток или способствуют их гибели, а в опухоли этот сдерживающий эффект подобных генов отсутствует, тем самым провоцируется рост опухолевых масс. Современная наука полагает, что для возникновения трансформированного клеточного клона необходимо как минимум пять-девять мутаций в разных онкогенах и антионкогенах.
— Эти мутации можно выявить с помощью генетического исследования?
— Да, конечно, можно. Спектр генетических повреждений в опухолях характеризуется удивительным многообразием. Например: амплификации (увеличение копийности генов), делеции, инсерции, транслокации, микромутации (точковые замены, микроделеции, микроинсерции) и так далее. Кроме того, в опухоли изменяются уровни экспрессии генов в результате аномального метилирования их промоторов.
Существует много методов, используемых для выявления мутаций в опухолевой ткани, и достаточно много ситуаций, когда это требуется. Выявление определённых мутаций иногда помогает поставить диагноз, определить лечебную тактику, прогноз и так далее. Наиболее часто для молекулярного тестирования используются технологии полимеразной цепной реакции (ПЦР) и секвенирования нового поколения (NGS, next generation sequencing). Обе технологии универсальны и используются для анализа любой генетической последовательности, а также многократно превосходят все другие технологии по своей чувствительности, специфичности и не сопряжены с риском получения «промежуточных», неинтерпретируемых результатов. Секвенирование экзома позволяет выявить все мутации в кодирующих последовательностях генома в каждой конкретной опухоли. Именно полногеномное секвенирование значительно расширяет возможности персонализированного подбора препаратов, предназначенных для специфического поражения мутированных онкобелков.

— Кому и чем могут помочь генетические исследования? Верно ли, что от генетического исследования может зависеть успех лечения? Кому стоит пройти генетическое исследование на мутации?
— Сфера медицинского применения ДНК- и РНК-тестов в современной онкологии постоянно расширяется. Сейчас это тестирование позволяет диагностировать наследственные опухолевые синдромы, выявить предиктивные мутации, осуществить анализ экспрессионных характеристик опухоли. Также совершенствуются технологии, которые позволяют уточнять диагноз опухолей с невыявленным первичным очагом, эффективно контролировать течение заболевания и изменения свойств опухоли (жидкостная биопсия), выполнять различные биологические тесты с опухолевыми клетками.
Индивидуализация лечения онкологического пациента во многих случаях напрямую зависит от результатов генетического тестирования. Эмпирический подход, сопряжённый со случайным перебором биологически активных химикатов, постепенно замещается научно-обоснованным, молекулярно-направленным поиском специфических противоопухолевых средств, направленных на активацию или инактивацию ключевых биохимических компонентов опухолевой трансформации.
Например, еще недавно клиническое деление всех первичных опухолей легкого на мелкоклеточный и немелкоклеточный рак было достаточным для определения стратегии лечения. Ситуация изменилась с открытием активирующих мутаций в гене, который кодирует рецептор эпидермального фактора роста — EGFR, сделавших этот онкогенный белок избирательной мишенью для воздействия препаратов ингибиторов EGFR. Мутации EGFR, как правило, встречаются у пациентов с аденокарциномой легкого. Тест на мутацию EGFR позволяет практически со 100%-й достоверностью отобрать тех больных, у которых гарантирован положительный результат применения гефитиниба, эрлотиниба или афатиниба.
— Может ли генетическое исследование помочь здоровому человеку предупредить рак или выявить его на ранней стадии?
— Вообще, бывают наследственные и ненаследственные опухоли. Наследственные опухолевые синдромы составляют незначительную долю от общего числа новообразований (около 1%), хотя для определённых локализаций (молочная железа, яичник, толстая кишка) их удельный вклад достигает более высоких показателей (5-20 %). Носительство наследуемой «раковой» мутации является причиной подобного заболевания. В этих случаях, в каждой клетке организма человека есть повреждение, которое передалось ему по наследству. Лица, имеющие такой генетический дефект, остаются практически здоровыми до определенного момента. В то же время они обладают фатально высоким риском возникновения опухолей (85-100%).
Генетическое исследование при подозрении на наследственный раковый синдром носит комплексный характер. Оно начинается со сбора онкологического анамнеза ‒ уделяется внимание случаям злокачественных заболеваний у кровных родственников. В результате составляются родословные, позволяющие заподозрить наследственную патологию. На заключительном этапе проводится анализ ДНК, что позволяет установить наличие в генотипе больного, а также членов его семьи, подозреваемые мутации.
— Какие виды мутаций ученые уже выявили? Существует ли для каждого вида таргетный препарат? Как именно работает таргетный препарат?
— Много разных видов мутаций при разных опухолях известны, но наибольший интерес представляют мутации в онкогенах, в частности, в рецепторных протеинкиназах, для блокировки которых разрабатываются специфические препараты. Мутации в протеинкиназах изменяют конформацию белковых молекул и, таким образом, формируют идеальное терапевтическое окно. Таргетный препарат избирательно воздействует на клетки опухоли, содержащие молекулярную мишень, и этим выгодно отличается от химиотерапии. Известно об успешном использовании ингибитора тирозинкиназы ALK – кризотиниба – у больных с ALK-транслоцированными карциномами легкого. Успешным оказалось и применение специфических ингибиторов мутированного белка BRAF – вемурафениба и дабрафениба для лечения больных меланомой. Другой пример: ген BRCA1 кодирует фермент репарации ДНК. BRCA1-дефицитные клетки демонстрируют неспособность эффективно удалять сшивки ДНК, индуцированные препаратами платины. В наследственных BRCA1-ассоциированных раках отмечается наибольшая эффективность цисплатина, т.к. в опухолевых клетках наблюдается соматическая утрата оставшегося BRCA1-аллеля, в то время как нормальные клетки носительниц мутаций BRCA1 сохраняют интактную копию данного гена. Этим обусловлено уникальное терапевтическое окно и это объясняет высокую эффективность цисплатина при лечении BRCA1-ассоциировнного рака молочной железы, яичника. Конечно, по разным причинам, не для всех мутаций есть такие препараты, но их спектр и количество неуклонно возрастает.
— Какие исследования, связанные с мутациями ДНК, сейчас проводятся в научной лаборатории молекулярной онкологии ФГБУ «НМИЦ онкологии им. Н.Н. Петрова»?
— В настоящее время проводятся исследования в двух направлениях: диагностика наследственных раковых синдромов и индивидуализация подбора лекарственных препаратов на основе молекулярных характеристик опухоли. Тем самым повышается клиническая эффективность применения дорогостоящих лекарственных препаратов, снижается частота и тяжесть побочных эффектов, и в некоторых случаях предотвращается неблагоприятный исход заболевания.
Читайте также: