Гиперэластическая кожа или синдром Элерса-Данлоса. Пигментная крапивница
Добавил пользователь Дмитрий К. Обновлено: 30.01.2026
Буллезный эпидермолиз. Болезнь Дарье или фолликулярный кератоз
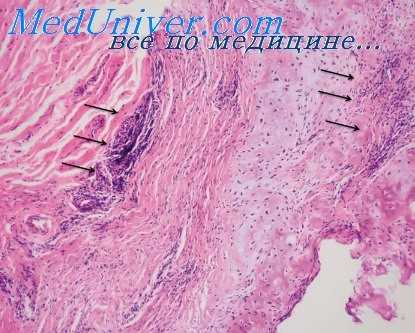
При этом заболевании образуются пузыри или пузырьки обычно на местах травмы, а иногда и без предшествующей травмы. Различают две формы дерматоза — простую (epidermolysis bullosa simplex) и дистрофическую (epidermolysis bullosa dystrophica). При простой форме пузыри заживают без образования рубца, слизистые оболочки и ногти поражаются редко, заболевание имеет тенденцию к самопроизвольному излечению или значительному улучшению ко времени полового созревания. При дистрофической форме высыпания заживают с образованием атрофических рубцов, часто наблюдаются поражения в полости рта и дистрофические изменения ногтей. Заболевание существует в течение всей жизни.
При буллезном эпидермолизе обычно бывают однокамерные пузыри и пузырьки, которые могут локализоваться в любом слое эпидермиса. При дистрофической форме они чаще всего располагаются под эпидермисом [Талипэн; Лэмб и Халперт (Tulipan; Lamb, Halpert)], при простой форме — под роговым слоем [Джонсон и Тест (Johnson, Test)]. При обеих формах в верхней части дермы имеется отек и хронический воспалительный инфильтрат различной интенсивности. Часто находят эозинофилы. При дистрофической форме в верхней части дермы могут обнаруживаться милиаподобные эпидермальные кисты (Талипэн).
Энгман и Мук (Engman, Mook) впервые описали при этом заболевании отсутствие эластических волокон в сосочковом и подсосочковом слоях пораженной и клинически здоровой кожи. Они полагали, что причиной заболевания является отсутствие эластических волокон. В дальнейшем некоторые авторы подтверждали эти данные, другие же [Гай и Аллен (Guy, Allen)] обнаруживали нормальную эластическую ткань. В последние годы установлено, что при простой форме буллезного эпидермолиза эластическая ткань нормальна (Джонсон и Тест), при дистрофической форме она имеет тенденцию исчезать в верхней части дермы (Талипэн; Лэмб и Халперт).
Возможно, однако, что отсутствие эластической ткани при дистрофической форме является не первичным, а вторичным симптомом, зависящим от разрушения ткани в результате патологического процесса.
Дифференциальный диагноз. Дифференцировать буллезный эпидермолиз от других пузырных заболеваний невозможно. Однако в его пользу свидетельствует наличие маленьких эпидермальных кист и отсутствие эластической ткани.
Болезнь Дарье или фолликулярный кератоз
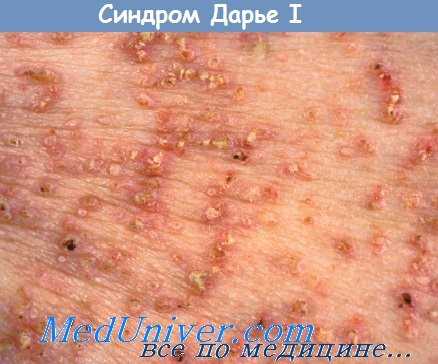
Заболевание характеризуется гиперкератотическими или покрытыми коркой узелками, которые, сливаясь друг с другом, могут образовывать бородавчатые разрастания. Выраженность этих высыпаний различна. Часто поражается слизистая оболочка полости рта, в отдельных случаях слизистая оболочка влагалища, гортани и глотки [Брюнауэр (Brunauer)].
Наличие папилломатоза в сочетании с гиперкератозом приводит к развитию гиперкератотических пробок, которые часто заполняют сально-волосяные фолликулы и, кроме того, обнаруживаются и вне фолликулов. Болезнь Дарье не является первичным фолликулярным заболеванием. Доказательством этому служит тот факт, что дерматоз может локализоваться на участках, лишенных фолликулярного аппарата, — на ладонях, подошвах, слизистой оболочке полости рта. Таким образом, термин «фолликулярный кератоз» ошибочен.
Сочетание папилломатоза и образования лакун приводит к своеобразной гистологической картине, которая может быть обозначена как вегетация. В участках вегетации растянутые и разветвленные сосочки покрыты одним, а иногда (несколькими слоями эпителиальных клеток.
Поражения слизистых оболочек аналогичны поражениям кожи (Брюнауэр).
Дифференциальный диагноз. Отличие болезни Дарье от семейной доброкачественной хронической пузырчатки см. ниже. Пролиферации типа villi («выступы») в область лакун могут напоминать картину при syringocystadenoma papilliferum [Бирман (Beerman)]. Однако при последнем заболевании villi ограничены двумя рядами эпителиальных клеток. Кроме того, отсутствует дискератоз, а в дерме имеются большие апокринные железы.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Гиперэластическая кожа или синдром Элерса-Данлоса. Пигментная крапивница
Этот синдром характеризуется: 1) усиленной эластичностью кожи; 2) чрезмерной гибкостью суставов; 3) ломкостью кожи с образованием рубцов и 4) развитием изюмоподобных псевдоопухолей. Псевдоопухоли мягкие и пигментированные; поверхность их морщиниста.
Гистопатология синдрома Элерса-Данлоса. Дегенеративные изменения обнаруживаются как в коллагеновой, так и в эластической ткани. Многие авторы полагают, что изменения коллагена являются первичными и доминирующими [Кортинг и Готтрон (Коrting, Gottron)]. Коллагеновые пучки становятся атрофичными; они расщепляются и отделяются друг от друга отеком.
Эластические волокна в некоторых случаях бывают нормальными, но в большинстве случаев они разрываются и имеют вид комков. Количество эластической ткани часто является увеличенным; по-видимому, однако, это увеличение является не истинным, а относительным (результат атрофии коллагена). Количество капилляров увеличено, их просветы расширены. Могут наблюдаться большие кистозные пространства, представляющие собой лимфангиэктатические полости (Кортинг и Готтрон).
Изюмоподобные псевдоопухоли, которые часто представляют собой часть данного синдрома, развиваются на участках травматических геморрагии и состоят из скопления гигантских клеток инородных тел [Рончезе (Ronchese)] или из пролиферированной соединительной ткани с большим количеством сосудов в ней.
Плотные подкожные узлы содержат обызвествленный некротический жир или слизистую массу, внедренную в толстую фиброзную капсулу [Джонсон и Фоллс (Falls)].

Пигментная крапивница
Это заболевание характеризуется большим количеством коричневых пятен, которые могут располагаться на всех участках кожного покрова. При трении пятен тупым предметом они превращаются в волдыри. В редких случаях поражение состоит из мягких узелков и бляшек.
Гистопатология пигментной крапивницы. Гистологическая картина характеризуется инфильтратом, состоящим преимущественно из тучных клеток. При пятнистом типе высыпаний тучные клетки расположены диффузно в верхней трети дермы. Некоторые тучные клетки имеют круглую или овальную форму, большинство же веретенообразной формы. Тучные клетки имеют тенденцию располагаться вокруг капилляров подсосочкового слоя и вблизи придатков кожи.
С возрастом количество тучных клеток уменьшается, поэтому при гистопатологическом исследовании пигментной крапивницы у взрослых больных они могут отсутствовать.
При узелковом типе пигментной крапивницы тучные клетки расположены компактно в виде опухолеподобных скоплений в верхней части дермы. Инфильтрат может через всю дерму проникать в подкожную жировую клетчатку.
Если тучные клетки расположены в виде густых агрегатов, они чаще имеют кубическую, чем веретенообразную форму; протоплазма их обильная и слегка эозинофильная. Вследствие своей формы и обилия протоплазмы они не напоминают никакие иные клетки, и диагноз может быть установлен без применения специальных окрасок.
Если биопсия производилась вскоре после того, как элементы подвергались трению, в препарате обнаруживается отек, большое количество эозинофилов и сморщивание тучных клеток со значительным уменьшением количества зерен в них, что указывает на выхождение зерен из тучных клеток [Дреннэн (Drennan)]. Иногда наблюдается даже разрушение и временное исчезновение тучных клеток, что может объяснить некоторые описанные в литературе типичные случаи пигментной крапивницы без обнаружения тучных клеток [Дреннэн и Бир (Веаге)].
Хронический дерматит. Контактный дерматит, монетовидная экзема
При хроническом дерматите часто бывает выраженный акантоз с удлинением эпидермальных отростков и гиперкератоз, перемежающийся с очагами паракератоза. Может быть небольшой межклеточный отек эпидермиса, но без образования пузырьков, в верхней части дермы — умеренно выраженный инфильтрат (преимущественно периваскулярный), состоящий из разнообразных клеток. В инфильтрате больше всего лимфоцитов, но количество эозинофилов, гистиоцитов и фибробластов также может быть значительным. Нейтрофилов нет.
Количество капилляров увеличено; стенки артериол и маленьких артерий могут быть утолщены. Для описанной гистологической картины хронического дерматита Заке, Миллер и Грей (Sash, Gray, 1496) применили термин «невродерматиче-ская реакция» вследствие гистологического сходства с диссе-минированным невродермитом (атопическим дерматитом) и ограниченным невродермитом (lichen simplex ehronicus). Однако аналогичная гистологическая картина может наблюдаться и при любом другом дерматозе, принадлежащем к группе дерматит-экзема.
Гистологическая картина хронического дерматита может быть при любом дерматозе, принадлежащем к группе дерматит-экзема. Кроме того, при многих заболеваниях, не относящихся к этой группе, могут иметься гистологические изменения (всегда или в отдельных случаях) не более специфичные, чем изменения при хроническом дерматите. К заболеваниям, при которых гистологические изменения всегда не специфичны и не отличаются от изменений при хроническом дерматите, относятся розовый лишай, парапсориаз, пеллагра и др.
При таких заболеваниях, как чешуйчатый лишай, красный плоский лишай и красная волчанка, гистологическая картина бывает специфичной в клинически типичных случаях, но она может быть не специфичной и не отличаться от картины хронического дерматита в клинически атипичных случаях.
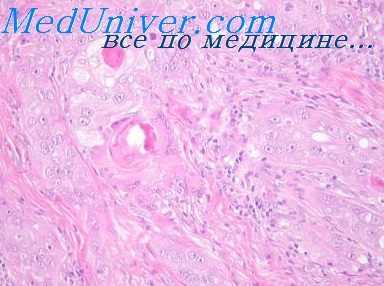
Когда при гистологическом исследовании обнаруживается картина хронического дерматита, всегда следует иметь в виду возможность ранней стадии грибовидного микоза. Часто бывает очень трудно гистологически диагностировать или отвергнуть возможность ранней стадии грибовидного микоза. В таких случаях следует искать атипичные гистиоциты, митотические фигуры, комкообразование ядер, кариорексис (распад ядер в «ядерную пыль») и микроабсцессы Потрие.
Однако некоторая атипичность гистиоцитов и отдельные митотические фигуры могут наблюдаться и при хроническом дерматите. В сомнительных случаях всегда рекомендуется сделать повторную биопсию.
Скажем несколько слов о гистологических изменениях при различных формах группы дерматит-экзема.
Контактный дерматит. Контактный дерматит может быть острым, подострым и хроническим. Гистологические изменения, описанные при остром, подостром и хроническом дерматите, в общем относятся и к контактному дерматиту. При остром контактном дерматите имеются многочисленные тесно расположенные большие и маленькие пузырьки.
При хроническом контактном дерматите отмечается неравномерный акантоз. Даже в этой стадии часто обнаруживаются маленькие внутриэпидермальные пузырьки (Закс, Миллер и Грэй, 1944; Миллер).
Монетовидная экзема (инфекционный экзематоидный дерматит). Высыпания, клинически выражающиеся «пузырьками с острие булавки», гистологически проявляются картиной подострого дерматита. В эпидермисе с умеренно выраженным акантозом имеются многочисленные рассеянные очаги внутриэпидермальных пузырьков (Закс, Миллер и Грэй, 1946). Как правило, спонгиоз вокруг пузырьков выражен слабо или отсутствует.
Атопический дерматит (диссеминированный невродермит). Гистологическая картина обычно характерна для хронического дерматита. Имеется акантоз; спонгиоз выражен слабо или совсем отсутствует. Количество эозинофилов в воспалительном инфильтрате может быть значительным (Буркхардт и Монтгомери).
- Вернуться в оглавление раздела "Дерматология"
Синдром Элерса - Данлоса
Синдром Элерса-Данлоса – наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур. В зависимости от клинического типа синдром Элерса-Данлоса может проявляться гипермобильностью суставов, необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием, птозом внутренних органов и пр. При диагностике синдрома Элерса-Данлоса учитываются клинические данные, результаты биопсии кожи и генотипирования; возможна пренатальная диагностика патологии. Лечение синдрома Элерса-Данлоса сводится к соблюдению щадящего режима, белковой диеты, симптоматической терапии.
Общие сведения
Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Сложность верификации и наличие легких форм затрудняет получение точных сведений об истинной распространенности синдрома Элерса-Данлоса; частота диагностированных среднетяжелых случаев составляет 1:5 000 новорожденным, тяжелых форм - 1:100 000.
Причины синдрома Элерса-Данлоса
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена. Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа; при VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена. VII тип обусловлен нарушением превращения проколлагена I типа в коллаген; X тип - патологией плазменного фибронектина, участвующего в организации межклеточного матрикса и т. п.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация синдрома Элерса-Данлоса
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса-Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно-доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса-Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса-Данлоса – имеет аутосомно-доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса-Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. ч. во внутренних органах), разрывам полых органов и сосудов (в т. ч. аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса-Данлоса – имеет Х-сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса-Данлоса - наследуется по аутосомно-рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелый кифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки и т. д.
VII тип синдрома Элерса-Данлоса (артроклазия) - наследуется как аутосомно-доминантно, так и аутосомно-рецессивно. Клиническую картину определяет низкий рост пациентов и гиперподвижность суставов, приводящая к частым привычным вывихам.
VIII тип синдрома Элерса-Данлоса – преимущественно наследуется аутосомно-доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса-Данлоса – характеризуется аутосомно-рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса-Данлоса – имеет аутосомно-доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
- классический (типы I и II)
- гипермобильный (тип III)
- сосудистый (тип IV)
- кифосколиоз (тип VI)
- артроклазия (тип VIIB)
- дермоспараксис (тип VIIC)
- недостаток тенасцина-X
Симптомы синдрома Элерса-Данлоса
Ввиду того, что подробная характеристика различных типов синдрома Элерса-Данлоса дана выше, в настоящем разделе обобщим основные проявления заболевания. Поскольку соединительная ткань присутствует практически во всех органах, проявления синдрома Элерса-Данлоса носят системный, генерализованный характер.
Ведущим в клинической картине является кожный синдром: гиперэластичность кожи, которая легко собирается в складку и оттягивается. На ощупь кожа бархатистая, нежная, слабо фиксированная с подлежащими тканями, морщинистая на ладонных и подошвенных поверхностях. Гиперэластичность кожи при синдроме Элерса-Данлоса отмечается с рождения или дошкольного возраста, с годами имеет тенденцию к снижению.
Кроме, гиперрастяжимости, характерна повышенная ранимость, хрупкость кожи, обнаруживающаяся в возрасте старше 2-3-х лет. Минимальная травматизация приводит к образованию длительно не заживающих ран, на месте которых спустя время формируются атрофичные или келоидные рубцы, псевдоопухоли.
Суставные проявления синдрома Элерса-Данлоса представлены гипермобильностью (разболтанностью) суставов, которая может носить локальный (например, переразгибание межфаланговых суставов) или генерализованный характер. Суставной синдром проявляется с началом ходьбы ребенка, что приводит к повторным подвывихам и вывихам. С возрастом гипермобильность суставов обычно уменьшается.
Со стороны сердечно-сосудистой системы у детей с синдромом Элерса-Данлоса нередко выявляются врожденные пороки сердца, пролапс митрального клапана, аневризмы сосудов головного мозга, варикоз. Отмечается склонность к кровотечениям - экхимозам, гематомам различной локализации, носовым, десневым, маточным, желудочно-кишечным кровотечениям.
Глазные проявления синдрома Элерса-Данлоса могут включать гиперэластичность кожи век, миопию, птоз, косоглазие, разрывы роговицы и глазного яблока при минимальных механических повреждениях, спонтанную отслойку сетчатки.
Изменения скелета при синдроме Элерса-Данлоса характеризуются воронкообразной или килевидной деформацией грудной клетки, сколиозом, кифозом, косолапостью, неправильным прикусом, частичной адентией. Висцеральные нарушения представлены птозом внутренних органов, пупочными, паховыми, диафрагмальными грыжами, рецидивирующим спонтанным пневмотораксом, дивертикулезом кишечника и др. Умственное развитие детей с синдромом Элерса-Данлоса обычно соответствует возрасту.
Диагностика синдрома Элерса-Данлоса
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз и т. д.).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
Лечение синдрома Элерса-Данлоса
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза, лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз синдрома Элерса-Данлоса
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Синдром Лайелла ( Злокачественная пузырчатка , Острый эпидермальный некролиз , Токсический эпидермальный некролиз )
Синдром Лайелла — это тяжелое полиэтиологическое заболевание аллергической природы, характеризующееся острым нарушением общего состояния пациента, буллезным поражением всего кожного покрова и слизистых. Быстрое развитие обезвоживания, токсическое поражение почек и других внутренних органов, присоединение инфекционного процесса часто приводят к летальному исходу заболевания. Диагностика синдрома Лайелла включает объективное обследование пациента, постоянный мониторинг данных коагулограммы, клинических и биохимических анализов крови и мочи. Лечение синдрома Лайелла включает проведение неотложных мероприятий, методы экстракорпорального очищения крови, инфузионную терапию, введение больших доз преднизолона, антибиотикотерапию, коррекцию водно-солевых нарушений и др.
МКБ-10
Синдром Лайелла (острый или токсический эпидермальный некролиз) относится к группе буллезных дерматитов. Свое основное название он получил в честь врача Лайелла, который в 1956 году впервые описал синдром как тяжелую форму токсикодермии. Клиническая картина синдрома Лайелла сходна с ожогом кожи II степени, в связи с чем заболевание называют ожоговым кожным синдромом. Еще одно распространенное название синдрома — злокачественная пузырчатка — обусловлено образованием на коже пузырей, подобных элементам пузырчатки.
Синдром Лайелла встречается в 0,3% случаев медикаментозных аллергий. После анафилактического шока он является самой тяжелой аллергической реакцией. Чаще всего синдром Лайелла наблюдается у людей молодого возраста и детей. Симптомы заболевания могут проявиться через пару часов или в течение недели после введения медикамента. По различным данным смертность при синдроме Лайелла составляет от 30% до 70%.
Причины
В зависимости от причины развития синдрома Лайелла современная дерматология выделяет 4 варианта заболевания.
- Первый представляет собой аллергическую реакцию на инфекционный процесс и чаще всего обусловлен золотистым стафилококком II группы. Как правило, он развивается у детей и отличается наиболее тяжелым течением.
- Второй — синдром Лайелла, который наблюдается в связи с применением лекарственных препаратов (сульфаниламидов, антибиотиков, противосудорожных лекарств, ацетилсалициловой кислоты, обезболивающих, противовоспалительных и противотуберкулезных средств). Наиболее часто развитие синдрома обусловлено одновременным приемом нескольких препаратов, одним из которых был сульфаниламид. В последние годы описаны случаи развития синдрома Лайелла на применение биологически активных добавок, витаминов, контрастных веществ для проведения рентгенографии и др.
- Третий вариант синдрома Лайелла составляют идеопатические случаи заболевания, причина возникновения которых остается невыясненной. Четвертый - синдром Лайелла, вызванный комбинированными причинами: инфекционными и лекарственными, развивается на фоне терапии инфекционного заболевания.
Патогенез
Большая роль в развитии синдрома Лайелла отводится генетически обусловленной предрасположенности организма к различным аллергическим реакциям. В анамнезе многих пациентов есть указания на аллергические заболевания: аллергический ринит, поллиноз, аллергический контактный дерматит, экзему, бронхиальную астму и др. У таких лиц из-за нарушения механизмов обезвреживания токсических продуктов обмена веществ происходит соединение введенного в организм лекарственного вещества с белком, который содержится в клетках эпидермиса. Это вновь образовавшееся вещество и является антигеном при синдроме Лайелла. Таким образом, иммунный ответ организма направлен не только на введенное лекарство, но и на кожу больного. Процесс напоминает реакцию отторжения трансплантата, в которой за трансплантат иммунная система принимает собственную кожу пациента.
В основе синдрома Лайелла лежит феномен Шварцмана-Санарелли — иммунологическая реакция, приводящая к нарушению регуляции распада белковых веществ и накоплению продуктов этого распада в организме. В результате происходит токсическое поражение органов и систем. Это нарушает работу обезвреживающих и выводящих токсины органов, что усугубляет интоксикацию, приводит к выраженным изменениям водно-солевого и электролитного баланса в организме. Данные процессы приводят к быстрому ухудшению состояния пациента при синдроме Лайелла и могут стать причиной летального исхода.
Симптомы
Синдром Лайелла начинается с внезапного и беспричинного повышения температуры тела до 39-40°С. За несколько часов на коже туловища, конечностей, лица, слизистой ротовой полости и гениталий появляются слегка отечные и болезненные эритематозные пятна различного размера. Они могут частично сливаться.
Через некоторое время (в среднем 12 часов) на участках внешне здоровой кожи начинает происходить отслаивание эпидермиса. При этом образуются тонкостенные вялые пузыри неправильной формы, величина которых варьирует от размеров лесного ореха до 10-15 см в диаметре. После вскрытия пузырей остаются большие эрозии, по периферии они покрыты обрывками покрышек пузырей. Эрозии окружены отечной и гиперемированной кожей. Они выделяют обильный серозно-кровянистый экссудат, что является причиной быстрого обезвоживания организма пациента.
При синдроме Лайелла отмечается характерный для пузырчатки симптом Никольского — отслаивание эпидермиса в ответ на незначительное поверхностное воздействие на кожу. В связи с отслойкой эпидермиса, на тех участках кожного покрова, которые подверглись сдавлению, трению или мацерации, эрозии формируются сразу, без образования пузырей.
Довольно быстро вся кожа пациента с синдромом Лайелла становится красной и резко болезненной при дотрагивании, ее внешний вид напоминает ожог кипятком II-III степени. Наблюдается характерный симптом «смоченного белья», когда кожа при прикосновении к ней легко сдвигается и сморщивается. В отдельных случаях синдрома Лайелла его основные проявления сопровождаются появлением мелкой петехиальной сыпи по всему телу больного. У детей заболевание обычно начинается с симптомов конъюнктивита и сочетается с инфекционным поражением кожи стафилококковой флорой.
Поражение слизистых при синдроме Лайелла проявляется образованием на них болезненных поверхностных дефектов, кровоточащих даже при незначительном травмировании. Процесс может затрагивать не только рот и губы, а и слизистую глаз, глотки, гортани, трахеи, бронхов, мочевого пузыря и уретры, желудка и кишечника.
Осложнения
Общее состояние пациентов с синдромом Лайелла прогрессивно ухудшается и за короткий период времени становится крайне тяжелым. Мучительная жажда, снижение потоотделения и продукции слюны являются признаками обезвоживания организма. Пациенты жалуются на выраженную головную боль, теряют ориентацию, становятся сонливыми. Наблюдается выпадение волос и ногтей. Обезвоживание приводит к сгущению крови и нарушению кровоснабжения внутренних органов. Наряду с токсическим поражением организма это приводит к нарушению работы печени, сердца, легких и почек. Развивается анурия и острая почечная недостаточность. Возможно присоединение вторичной инфекции.
Диагностика
Клинический анализ крови при синдроме Лайелла указывает на воспалительный процесс. Наблюдается повышение СОЭ, лейкоцитоз с появлением незрелых форм. Снижение или полное отсутствие эозинофилов в анализе крови является диагностическим признаком, позволяющим отличить синдром Лайелла от других аллергических состояний. Данные коагулограммы указывают на повышенную свертываемость крови. Анализ мочи и биохимический анализ крови позволяют выявить нарушения, происходящие со стороны почек, и осуществлять мониторинг состояния организма в процессе лечения.
Важной задачей является определение медикамента, который привел к развитию синдрома Лайелла, ведь его повторное применение в процессе лечения может быть губительным для пациента. Выявить провоцирующее вещество помогает проведение иммунологических тестов. На провоцирующий препарат указывает быстрое размножение иммунных клеток, возникающее в ответ на его введение в образец крови пациента.
Биопсия кожи и гистологическое изучение полученного образца у больного синдромом Лайелла обнаруживает полную гибель клеток поверхностного слоя эпидермиса. В более глубоких слоях наблюдается образование крупных пузырей, отечность и скопления иммунных клеток с наибольшей концентрацией в области кожных сосудов.
Синдром Лайелла дифференцируют с другими острыми дерматитами, сопровождающимися образованием пузырей: актиническим дерматитом, пузырчаткой, контактным дерматитом, синдромом Стивенса-Джонсона, буллезным эпидермолизом, герпетиформным дерматитом Дюринга, простым герпесом.
Лечение синдрома Лайелла
Лечение пациентов с токсическим эпидермальным некролизом проводится в реанимационном отделении и включает целый комплекс неотложных мероприятий. При этом, учитывая токсико-аллергический характер заболевания, применение лекарственных препаратов должно проводиться со строгим учетом показаний и противопоказаний.
Терапия синдрома Лайелла осуществляется инъекционным введением больших доз кортикостероидов (преднизолон). При улучшении пациент переводится на прием препарата в таблетированной форме с постепенным понижением его дозы. Применение методов экстракорпоральной гемокоррекции (плазмаферез, гемосорбция) позволяет производить очищение крови от образующихся при синдроме Лайелла токсических веществ. Постоянная инфузионная терапия (физ. раствор, декстран, солевые растворы) направлена на борьбу с обезвоживанием и нормализацию водно-солевого баланса. Она проводится при строгом контроле объема выделяемой пациентом мочи.
В комплексной терапии синдрома Лайелла применяют медикаменты, поддерживающие работоспособность почек и печени; ингибиторы ферментов, участвующих в разрушении тканей; минеральные вещества (калий, кальций и магний); препараты, снижающие свертываемость; мочегонные средства; антибиотики широкого спектра.
Местное лечение синдрома Лайелла включает применение аэрозолей с кортикостероидами, влажно-высыхающих повязок, антибактериальных примочек. Она проводится в соответствии с принципами обработки ожогов. Для профилактики инфицирования при синдроме Лайелла необходимо несколько раз в сутки проводить смену нательного белья на стерильное, осуществлять обработку не только кожи, но и слизистых. Учитывая выраженную болезненность, местная терапия должна проводиться при соответствующем обезболивании. При необходимости проведение перевязок осуществляется под наркозом.
Прогноз
Прогноз заболевания определяется характером его течения. В связи с этим выделяют 3 варианта течения синдрома Лайелла: молниеносное с летальным исходом, острое с возможным летальным исходом при присоединении инфекционного процесса и благоприятное, обычно разрешающееся спустя 7-10 дней. Ранее начало лечебных мероприятий и их тщательное проведение улучшают прогноз заболевания.
1. Синдром Лайелла как редкое осложнение медикаментозной терапии (клинические случаи)/ Тезяева С.А., Млинник Р.А., Дегтярева С.Ф., Вагапова Т.В, Никольский В.О.// МедиАл. - 2015 - №2 (16).ь
2. К проблеме лечения синдрома Лайелла: вопросы дискутабельного характера/ Владыка А.С. и др.// Украинский журнал дерматологии, венерологии и косметологии. - 2007.
3. Синдром Лайелла (клиника, диагностика, современные методы лечения)/ Чичерина Е.Н. Малых Св. Акшенцева М.В.// Вятский медицинский вестник. - 2008.
Читайте также: