Хромосомные аномалии при лимфогранулематозе - прогноз
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Лимфома неходжкенская — гетерогенная группа заболеваний, характеризующаяся неопластической пролиферацией незрелых лимфоидных клеток, накапливающихся вне костного мозга.
Лимфосаркоматоз (Кундрата болезнь) — генерализованная форма неходжкенской лимфомы, характеризующаяся множественным поражением лимфатических узлов, а в последующем — поражением печени и селезёнки.
Частота. Ежегодно в США диагноз неходжкенской лимфомы устанавливают примерно у 35000 больных.
Патогистологическая классификация. Известно множество гистологических классификаций заболевания. Для устранения противоречий между ними в 1982 г. принята классификация Национального Института Рака: • Лимфома низкой степени злокачественности •• Мелкоклеточная лимфоцитарная •• Преимущественно фолликулярная (мелкие клетки с расщеплёнными ядрами) •• Фолликулярная — смешанного типа (мелкие клетки с расщеплёнными ядрами и крупные клетки) • Лимфома средней степени злокачественности •• Преимущественно фолликулярная крупноклеточная •• Диффузная мелкоклеточная с расщеплёнными ядрами •• Диффузная смешанная (мелко- и крупноклеточная) •• Диффузная крупноклеточная • Лимфома высокой степени злокачественности •• Крупноклеточная •• Лимфобластная с изогнутыми ядрами •• Мелкоклеточная с нерасщеплёнными ядрами (Беркетта).
Этиология • Иммунодефициты • Длительный приём иммунодепрессантов (например, после пересадки почки или сердца) • Вирус Эпстайна–Барр связан с развитием лимфом Беркетта • Цитогенетические аномалии (например, хромосомные транслокации).
Клиническая картина • Пролиферативный синдром: лимфаденопатия (увеличение поражённых лимфатических узлов); опухолевый синдром: увеличение печени, селезёнки • Интоксикационный синдром: лихорадка, повышенная утомляемость, снижение массы тела и ночные поты • Клинические проявления зависят от локализации лимфосаркомы (кишечная непроходимость при абдоминальной локализации; синдром сдавления трахеи при поражении внутригрудных лимфатических узлов).
Стадии заболевания и диагностика • Принципы стадирования аналогичны таковым при лимфогранулематозе. 4-я стадия заболевания выставляется при вовлечении в патологический процесс костного мозга (лейкемизация) и ЦНС • Установление стадии •• Биопсия лимфатического узла и анализ биопсийного материала •• Гематологическое исследование, в т.ч. подсчёт лейкоцитарной формулы, тромбоцитов, определение содержания мочевой кислоты. Электрофорез белков крови позволяет исключить гипогаммаглобулинемию и/или болезнь тяжёлых цепей •• Сбор полного анамнеза и врачебный осмотр с акцентом на все группы лимфатических узлов (в первую очередь, кольцо фон Вальдейера–Пирогова), а также на размеры печени и селезёнки •• Двусторонняя биопсия и аспирация костного мозга •• Радиологические исследования — рентгенография органов грудной клетки, брюшной полости и таза, реже — двусторонняя лимфангиография нижних конечностей и таза •• Другие процедуры — диагностическая лапаротомия, сцинтиграфия или рентгенография костей, эндоскопия и биопсия печени.
ЛЕЧЕНИЕ обычно комбинированное. Как и при лечении лейкозов используют различные протоколы химиотерапии.
• Химиотерапия •• Лимфомы I и II стадии промежуточной и высокой степени злокачественности часто хорошо реагируют на комбинированную химиотерапию (большие дозы циклофосфамида с метотрексатом, винкристином и часто с доксорубицином) с облучением или без лучевой терапии (излечение наблюдают в 80–90% случаев) •• При поражении ЦНС цитостатики вводят эндолюмбально или в желудочки головного мозга.
• Облучение. Неходжкенские лимфомы чрезвычайно радиочувствительны •• При локализованном процессе облучение должно быть направлено на поражённую область (в дозе 40 Гр) •• При диссеминированной лимфоме облучение имеет паллиативный эффект, а также усиливает лечебный эффект химиотерапии •• I стадия вялотекущих лимфом. Длительное наблюдение за больными с локализованными I и II стадиями лимфомы низкой степени злокачественности, получившими общее облучение лимфатических узлов, выявило наличие 10-летнего безрецидивного периода в 50% случаев (особенно у молодых пациентов).
Особенности у детей
• Преобладающий возраст — 5–9 лет, соотношение мальчики/девочки — 2–2,5/1.
• Особенности течения •• Быстрое прогрессирование опухоли •• Преобладание экстранодальной локализации •• Первично-генерализованная опухоль.
• Локализация •• В-клеточные лимфомы — кишечник (35%), носоглотка (20%) •• Т-клеточные лимфомы — средостение (25%), периферические лимфатические узлы (15%).
• Лечение •• Основной метод — комбинированная полихимиотерапия •• Лучевую терапию применяют только при поражении ЦНС (местно).
• Течение и прогноз. 5-летняя выживаемость при лечении достигает 80%.
Синонимы • Лимфосаркома • Лимфобластома • Лимфома злокачественная.
МКБ-10 • C82 Фолликулярная [нодулярная] неходжкинская лимфома • C83 Диффузная неходжкинская лимфома
Примечание. Повышение заболеваемости у пожилых людей происходит за счёт диффузной крупноклеточной лимфомы.
Код вставки на сайт
Лимфома неходжкенская
Лимфома неходжкенская — гетерогенная группа заболеваний, характеризующаяся неопластической пролиферацией незрелых лимфоидных клеток, накапливающихся вне костного мозга.
Лимфосаркоматоз (Кундрата болезнь) — генерализованная форма неходжкенской лимфомы, характеризующаяся множественным поражением лимфатических узлов, а в последующем — поражением печени и селезёнки.
Частота. Ежегодно в США диагноз неходжкенской лимфомы устанавливают примерно у 35000 больных.
Патогистологическая классификация. Известно множество гистологических классификаций заболевания. Для устранения противоречий между ними в 1982 г. принята классификация Национального Института Рака: • Лимфома низкой степени злокачественности •• Мелкоклеточная лимфоцитарная •• Преимущественно фолликулярная (мелкие клетки с расщеплёнными ядрами) •• Фолликулярная — смешанного типа (мелкие клетки с расщеплёнными ядрами и крупные клетки) • Лимфома средней степени злокачественности •• Преимущественно фолликулярная крупноклеточная •• Диффузная мелкоклеточная с расщеплёнными ядрами •• Диффузная смешанная (мелко- и крупноклеточная) •• Диффузная крупноклеточная • Лимфома высокой степени злокачественности •• Крупноклеточная •• Лимфобластная с изогнутыми ядрами •• Мелкоклеточная с нерасщеплёнными ядрами (Беркетта).
Этиология • Иммунодефициты • Длительный приём иммунодепрессантов (например, после пересадки почки или сердца) • Вирус Эпстайна–Барр связан с развитием лимфом Беркетта • Цитогенетические аномалии (например, хромосомные транслокации).
Клиническая картина • Пролиферативный синдром: лимфаденопатия (увеличение поражённых лимфатических узлов); опухолевый синдром: увеличение печени, селезёнки • Интоксикационный синдром: лихорадка, повышенная утомляемость, снижение массы тела и ночные поты • Клинические проявления зависят от локализации лимфосаркомы (кишечная непроходимость при абдоминальной локализации; синдром сдавления трахеи при поражении внутригрудных лимфатических узлов).
Стадии заболевания и диагностика • Принципы стадирования аналогичны таковым при лимфогранулематозе. 4-я стадия заболевания выставляется при вовлечении в патологический процесс костного мозга (лейкемизация) и ЦНС • Установление стадии •• Биопсия лимфатического узла и анализ биопсийного материала •• Гематологическое исследование, в т.ч. подсчёт лейкоцитарной формулы, тромбоцитов, определение содержания мочевой кислоты. Электрофорез белков крови позволяет исключить гипогаммаглобулинемию и/или болезнь тяжёлых цепей •• Сбор полного анамнеза и врачебный осмотр с акцентом на все группы лимфатических узлов (в первую очередь, кольцо фон Вальдейера–Пирогова), а также на размеры печени и селезёнки •• Двусторонняя биопсия и аспирация костного мозга •• Радиологические исследования — рентгенография органов грудной клетки, брюшной полости и таза, реже — двусторонняя лимфангиография нижних конечностей и таза •• Другие процедуры — диагностическая лапаротомия, сцинтиграфия или рентгенография костей, эндоскопия и биопсия печени.
ЛЕЧЕНИЕ обычно комбинированное. Как и при лечении лейкозов используют различные протоколы химиотерапии.
• Химиотерапия •• Лимфомы I и II стадии промежуточной и высокой степени злокачественности часто хорошо реагируют на комбинированную химиотерапию (большие дозы циклофосфамида с метотрексатом, винкристином и часто с доксорубицином) с облучением или без лучевой терапии (излечение наблюдают в 80–90% случаев) •• При поражении ЦНС цитостатики вводят эндолюмбально или в желудочки головного мозга.
• Облучение. Неходжкенские лимфомы чрезвычайно радиочувствительны •• При локализованном процессе облучение должно быть направлено на поражённую область (в дозе 40 Гр) •• При диссеминированной лимфоме облучение имеет паллиативный эффект, а также усиливает лечебный эффект химиотерапии •• I стадия вялотекущих лимфом. Длительное наблюдение за больными с локализованными I и II стадиями лимфомы низкой степени злокачественности, получившими общее облучение лимфатических узлов, выявило наличие 10-летнего безрецидивного периода в 50% случаев (особенно у молодых пациентов).
Особенности у детей
• Преобладающий возраст — 5–9 лет, соотношение мальчики/девочки — 2–2,5/1.
• Особенности течения •• Быстрое прогрессирование опухоли •• Преобладание экстранодальной локализации •• Первично-генерализованная опухоль.
• Локализация •• В-клеточные лимфомы — кишечник (35%), носоглотка (20%) •• Т-клеточные лимфомы — средостение (25%), периферические лимфатические узлы (15%).
• Лечение •• Основной метод — комбинированная полихимиотерапия •• Лучевую терапию применяют только при поражении ЦНС (местно).
• Течение и прогноз. 5-летняя выживаемость при лечении достигает 80%.
Синонимы • Лимфосаркома • Лимфобластома • Лимфома злокачественная.
МКБ-10 • C82 Фолликулярная [нодулярная] неходжкинская лимфома • C83 Диффузная неходжкинская лимфома
Примечание. Повышение заболеваемости у пожилых людей происходит за счёт диффузной крупноклеточной лимфомы.
Хромосомные аномалии при лимфогранулематозе - прогноз
Хромосомные аномалии при лимфогранулематозе - прогноз
Лимфогранулематоз (Hodgkin's lymphoma) представляет собою клональное новообразование из малигнизированных В-клеток терминального центра лимфатических узлов с перестроенным (реаранжированным) генами Н-цепей иммуноглобулинов (IgH) и приобретших соматические мутации («мутированный» VH-статус).
В большинстве случаев опухолевые клетки составляют лишь небольшую фракцию в каждом образце из пораженной ткани, поступающем для морфологического и/или цитогенетического анализа. Это многоядерные клетки Березовского—Штернберга или одноядерные клетки Ходжкина (в англоязычной литературе соответственно Reed-Sternberg cells и Hodgkin's cells).
При цитогенетическом анализе в пораженных лимфатических узлах выявляются клоны полиплоидных клеток с множественными числовыми изменениями кариотипа и очень сложными структурными перестройками хромосом. Происхождение возникающих хромосомных маркеров трудно установить без дополнительных молекулярно-генетических методик. Доля клеток с хромосомными нарушениями обычно менее 50 %, большинство метафазных пластинок нормальные.
Показано, что клоны цитогенетически измененных клеток — это клоны CD30-позитивных элементов. Как правило, это клетки Ходжкина и Березовского — Штернберга.
Лимфогранулематоз сравнительно мало исследовали с помощью стандартного цитогенетического метода, поскольку, во-первых, опухолевых клеток значительно меньше, чем нормальных, а во-вторых, из этой опухоли трудно получить пригодные для анализа хромосомные препараты. Имеется несколько оригинальных публикаций и обзоров.
Характерные (повторяющиеся) аномалии кариотипа выявлены главным образом с помощью таких молекулярно-генетических подходов, как FISH, FICTION и сравнительная геномная гибридизация (CGH).
Некоторые исследователи отмечают своеобразие нарушений кариотипа при лимфогранулематозе: характерны моносомии и делеции хромосомы 13, а также перестройки, затрагивающие районы 2р25, 12р11-13, 13р11-13, 14р11, 15pll-13, 20q12—13. В других работах, выполненных теми же методами, установлен несколько отличающийся спектр нарушений кариотипа,
Так, по данным S. Joos и соавт., наиболее часто наблюдается избыток хромосомного материала районов 2р, 9р и 12q, причем амплифицированы участки 4p16, 4q23-24, а также 9р23-24.

Лимфогранулематоз. Частота появления дополнительного материала и утрат различных участков генома по данным метода сравнительной геномной гибридизации.
Результаты обследования 41 пациента нанесены на схему нормального кариотипа — идеограмму. Каждому случаю соответствует вертикальная линия. Справа — приобретения, слева — утраты. Сравнительно часто наблюдается дополнительный материал в области короткого плеча хромосом 2, 9, 17, X и длинного плеча хромосом 9, 12, 17, X, реже — утраты в области короткого плеча хромосом 17 и X, а также длинного плеча хромосом 4, 13 и X.
Приведем частоты различных нарушений (утрат и избытка хромосом) из работы S. Joos и соавт., в которой также использовалась CGH. Повторяющиеся утраты генетического материала обнаружены авторами только в длинном плече хромосомы 13 (22 % случаев). Напомним, что эти нарушения характерны также для таких разных опухолей из зрелых В-клеток, как множественная миелома и ХЛЛ; в первом случае они имеют отрицательное прогностическое значение, а во втором ассоциированы с хорошим прогнозом.
Причина таких различий остается невыясненной. Мы не нашли в литературе данных о прогностическом значении делеций 13q при лимфогранулематозе. Кроме делеций 13q, по данным Joos и соавт., для лимфогранулематоза характерны перестройки, приводящие к избытку хромосомного материала. Так, в хромосомном районе 2р частота нарушений составляет 64%, 12q — 37%, 17р — 27 %, 9р-24%, 16р — 24 %, 17q — 20 %, 20q-20%.
Большинство исследователей считают, что для лимфогранулематоза характерны перестройки короткого плеча хромосомы 2 (2р). Так, в одной из недавних работ приводятся результаты исследования лимфатических узлов 44 больных, причем изучались два гена, расположенные в области 2р: BCL1A и REL. Оба гена являются транскрипционными факторами и оба уже были «замечены как соучастники» в генезе лимфом.
Авторам работы удалось обнаружить амплификацию хромосомного района 2р13 у 55 % больных лимфогранулематозом, причем были выявлены частые изменения именно гена REL: в одних случаях это была амплификация, в других — участие в транслокациях с различными хромосомами.
Специфические изменения кариотипа, характерные только для лимфогранулематоза, отличающие его от других опухолей, не выявлены.
Применение молекулярно-генетических методик позволило обнаружить изменения генов ТР53, IкВа и CD95. Частота случаев с этими мутациями варьирует. В клетках Березовского—Штернберга и Ходжкина нередко наблюдаются мутации генов JAK2, с-REL, MDM2, которые считают вовлеченными в процесс малигнизации.
Фактические данные однозначно свидельствуют в пользу практической значимости хромосомного анализа при гемобластозах. Показано, что у хромосомного анализа, как и у любого лабораторного метода, есть ограничения: прежде всего не очень высокая чувствительность и разрешающая способность по сравнению с молекулярно-генетическими методиками. Наибольшая точность диагностики достигается при рациональном сочетании классического хромосомного анализа с различными молекулярно-генетическими, цитохимическими и иммунофенотипическими исследованиями.
Онкологическая цитогенетика — уже зрелая, но постоянно развивающаяся область науки, в которой обязательно будут получены новые знания:
1) будут определены новые неслучайные изменения кариотипа; важные для диагностики;
2) будут выявлены новые хромосомные участки, в которых локализованы еще неизвестные гены, вовлеченные в онкогенез;
3) будут выявлены новые прогностические маркеры.
Остается еще ряд нерешенных вопросов, в частности:
1) неясно практическое значение многих редких цитогенетических и молекулярно-генетических аномалий, повторяющихся при разных вариантах лейкозов и лимфом;
2) неясна роль вторичных (дополнительных) изменений кариотипа в прогрессии заболевания и значение этих изменений в прогнозировании ответа на лечение;
3) нет пока рациональных протоколов молекулярного мониторинга МРБ, адаптированных к отдельным группам гемобластозов и программам их лечения.
Эти и другие неясные вопросы онкологической цитогенетики могут быть решены только большими многоцентровыми исследованиями.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Острые лейкозы
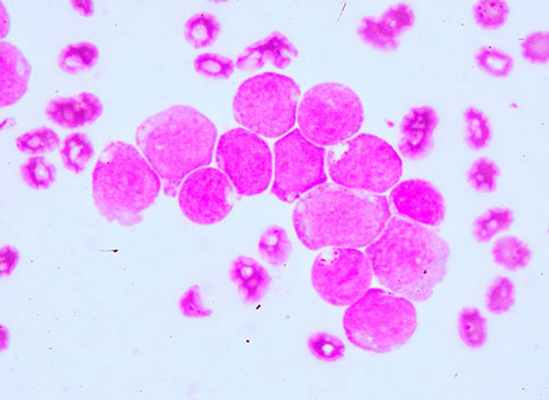
Лейкозы острые — это гетерогенная группа опухолевых заболеваний кроветворной системы, которые начинаются в костном мозге и характеризуются накоплением недифференцированных (бластных) клеток и подавлением нормальных ростков кроветворения. Заболеваемость острыми лейкозами — в среднем 3—5 первичных случаев на 100 000 человек в год. Острые лейкозы распространены повсеместно в разных странах, встречаются во всех возрастных группах, но у детей их удельный вес наибольший среди всех гемобластозов. Мужчины и женщины заболевают с равной частотой.
Классификация
Острые лейкозы классифицируют в зависимости от цитохимических и иммунофенотипических особенностей опухолевых клеток, выделяют:
-острые лимфобластные
-острые нелимфобластные лейкозы.
Современная классификация острых лейкозов предложена группой французских, американских и британских гематологов - FAB-классификация.
Предлейкоз — это доклиническая стадия острого лейкоза, при которой ограниченное количество бластов еще не вызывает явных клинических расстройств (МДС - миелодиспластические синдромы).
Клиника
Клиника развернутой стадии острого лейкоза: характерны нарастающая «беспричинная» слабость, недомогание, повышение Т°. Довольно часто геморрагический синдром: кровоточивость слизистых оболочек (десневые, носовые, маточные, кишечные кровотечения), петехиальная сыпь на коже, в первую очередь голеней, реже наблюдается гематурия и еще реже — кровоизлияние в мозг. Геморрагический синдром - нередко самое раннее проявление острого лейкоза.
При острых миеломнобластных лейкозах характерна гиперплазия десен, из-за которой больные часто впервые обращаются к стоматологу. Весьма характерны язвенно-некротические поражения слизистых оболочек ротовой полости (стоматит), глотки (ангина) и кишечника - некротическая энтеропатия, парапроктит и др.. что объясняется снижением уровня гранулоцитов крови. Начальные симптомы некротической энтеропатии - плеск и урчание при пальпации илеоцекальной зоны, кашицеобразный стул, легкое вздутие живота. В крови в большинстве случаев выявляются бластные клетки - уродливые, с деформированными нуклеолами, двухядерные; обычно много митозов. В костном мозге содержание бластов достигает иногда 95-99%. За счет бластов общий лейкоцитоз в крови иногда очень высок, изредка он в норме; известны и лейкопенические варианты дебюта острого лейкоза. При кариологическом исследовании в бластных клетках обнаруживаются хромосомные аномалии необычное число хромосом, аберрации и т.д.). Анемия при остром лейкозе нередко наступает позднее, чем тромбоцитопения и агранулоцитоз, она объясняется как кровотечениями, так и бластозом в костном мозге. Симптомы анемии весьма типичны для острого лейкоза; обычно малокровие носит нормохромный или гипохромный характер.
Первыми проявлениями острого лейкоза может быть также «беспричинный» субфебрилитет, изолированное увеличение лимфоузла, увеличение селезенки, лейкопения, тромбоцитопения, упорные боли в суставах и др. Увеличение лимфоузлов, печени и селезенки в начале развернутой стадии встречается не всегда, но с течением времени развивается у многих больных острым лейкозом. Болезненность костей выявляется лишь при большой массе лейкозных клеток, т.е. в уже запущенных случаях острого лейкоза. Опыт показывает, что даже при высоком бластозе в крови и костном мозге у пациента может сохраняться достаточный уровень гемоглобина, тромбоцитов и нейтрофилов, и общее состояние остается удовлетворительным (пациент даже отказывается от дальнейшего обследования, утверждая, что он чувствует себя нормально). Обычно это наблюдается при остром лимфобластном лейкозе (изменения в крови выявляются при периодическом осмотре, диспансеризации и т.д.). Задача врача в этих случаях — убедить пациента и его родственников в неотложной необходимости обследования. В других случаях даже небольшой бластоз вызывает выраженную слабость, глубокую тромбоцитопению, анемию и гранулоцитопению. что сразу влечет клинические осложнения. При всех вариантах острого лейкоза наблюдаются те или иные нарушения в свертывающей системе крови, вплоть до ДВС-синдрома, особенно при большом содержании бластов, тем более атипичных промиелоцитов в крови.
ДВС-синдром влечет за собой кровоточивость, нередко летальную.
Острый лимфобластный лейкоз
Острый лимфобластный лейкоз (ОЛЛ) - злокачественная опухоль крови, возникающая в результате мутации на уровне клетки-предшественницы лимфопоэза. Этот тип лейкоза является самым частым онкогематологическим заболеванием у детей (они составляют около 85% всех случаев острых лейкозов). У взрослых острый лимфобластный лейкоз наблюдается в 25% случаев всех острых лейкозов. Учашение заболеваемости острыми лимфобластными лейкозами отмечается у детей в возрасте 2—4 года, затем у лиц в возрасте 50—60 лет.
Классификация: на основе светооптических характеристик лейкозных клеток выделяют три основных варианта ОЛЛ: LI, L2 и L3. У детей чаше наблюдается вариант LI, у взрослых - форма L2.
При иммунофенотипировании различают четыре варианта опухоли:
- пре-В-клеточный вариант,
- ранний пре-В-клеточный,
- Т-клеточный (около 15% всех случаев),
- зрелый В-клеточный (весьма редкий).
После уточнения варианта ОЛЛ, применяют наиболее подходящую лечебную схему и оценивают прогноз для жизни. Так, у детей с ранним пре-В-клеточным вариантом ОЛЛ прогноз достоверно лучше; зрелые В-клеточные острые лимфобластные лейкозы менее благоприятны прогностически, особенно у взрослых. Цитогенетические исследования обнаруживают хромосомные аберрации более чем у половины больных острыми лимфобластными лейкозами. Наиболее серьезный риск-фактор - наличие «филадельфийской» хромосомы. У детей с острым лимфобластным лейкозом эта транслокация встречается в 7% случаев, у взрослых — в 30% случаев. Ухудшает прогноз наличие высокого уровня сывороточной лактатдегидрогеназы в момент выявления ОЛЛ. Один из ведущих риск-факторов — это высокий лейкоцитоз. Отметим, что у больных старше 50 лет ремиссии ОЛЛ удается добиться реже, чем у пациентов более молодого возраста.
Клиническая симптоматика при острых лимфобластных лейкозах: в начале заболевания обычно жалоб нет, или они весьма скудные — умеренная слабость и утомляемость, иногда бледность кожных покровов. В развернутой стадии ОЛЛ может наблюдаться лихорадка, геморрагические явления, малокровие и др. Однако анемизация и геморрагический диатез выражены значительно менее, чем при острых нелимфобластных лейкозах. При острых лимфобластных лейкозах увеличение печени наблюдается не всегда. У молодых больных острыми лимфобластными лейкозами возможна умеренная диффузная лимфоаденопатия. Первыми проявлениями острого лейкоза может быть также «беспричинный» субфебрилитет, бледность кожи и слизистых оболочек, кровоточивость десен, обильные менструации, изолированное увеличение лимфоузла, упорные боли в суставах и др. Почти всегда наблюдается слабость, утомляемость, потливость. Нередко отмечаются рецидивирующие ЛОР-инфекции, герпес. Болезненность костей выявляется лишь при большой массе лейкозных клеток, т.е. в уже запущенных случаях острого лейкоза. Даже при высоком бластозе в крови и костном мозге у пациента может сохраняться достаточный уровень гемоглобина, тромбоцитов и нейтрофилов. и общее состояние остается удовлетворительным.
Диагностика: поскольку специфичных симптомов при острых лейкозах не существует, диагноз устанавливают только после морфологического исследования крови и костного мозга. Появление даже единичных бластных клеток в периферической крови (гемограмма) требует безотлагательного исследования костного мозга (стернальная пункция).
Принципы лечения: госпитализация в гематологический стационар.
Химиотерапия, поспешно начатая в амбулаторных условиях, полностью исключает возможность последующего излечения пациента даже в условиях гематологического центра. Не рекомендуется начинать цитостатическое лечение в амбулаторных условиях, иногда однократное введение цитостатиков или глюкокортикостероидов до неузнаваемости меняет клиническую и гематологическую картину, и в дальнейшем не позволяет поставить правильный диагноз.
Лечение острых лейкозов предполагает:
- индукцию ремиссии,
- консолидацию полученной ремиссии,
- поддерживающую терапию,
- а также профилактику нейролейкемии.
Применяют патогенетическое лечение острого лейкоза с целью достижения полной клинико-гематологической ремиссии. В настоящее время общепринят метод интенсивной комбинированной длительной цитостатической терапии острого лейкоза (тотальная терапия).
Нелимфобластные острые лейкозы
Нелимфобластные острые лейкозы - эти варианты лейкозов возникают в результате мутации одной из миелоидных клеток-предшественников. Преобладают среди лейкозов взрослых.
В группе острых миелоидных лейкозов выделяют следующие формы заболевания:
МО-острый миелобластный лейкоз, при котором бластные клетки имеют минимальные признаки дифференцировки;
M1-2—острый миелобластный лейкоз с наличием клеток разной степени зрелости;
МЗ—острый промиелоцитарный лейкоз;
М4- острый миеломонобластный лейкоз;
М5—острый монобластный лейкоз;
М6—острый эритролейкоз;
М7- острый мегакариобластный лейкоз.
Острый миелобластный (ОМЛ) и миеломонобластный лейкозы (ОММЛ) встречаются чаще всего: среди нелимфобластных острых лейкозов взрослых эти формы составляют ок. 80%. Возрастные пики острого миелобластного и миеломонобластного лейкоза наблюдаются в возрасте до 1-2 лет, затем - в 38 лет (ОМЛ) и в 50 лет (ОММЛ). Клиника: характерно острое начало болезни с высокой лихорадкой, некрозами в горле (при глубокой первичной гранулоцитопении); часто бывают боли в костях, малокровие, геморрагические явления, которые обусловлены не только тромбоцитопенией, но и ДВС-синдромом. Наблюдается кровоточивость слизистых оболочек (десневые, носовые, маточные, кишечные кровотечения), петехиальная сыпь на коже, в первую очередь голеней, реже наблюдается гематурия и еще реже — кровоизлияние в мозг. Геморрагический синдром - нередко самое раннее проявление острого нелимфобластного лейкоза. Увеличение селезенки при остром миелобластном и миеломонобластном лейкозе умеренное. Весьма характерны язвенно-некротические поражения слизистых оболочек ротовой полости (стоматит, гиперпластический гингивит с кровоточивостью), глотки (ангина) и кишечника - некротическая энтеропатия, парапроктит и др., что объясняется снижением уровня гранулоцитов крови. Начальные симптомы некротической энтеропатии: плеск и урчание при пальпации илеоцекальной зоны, кашицеобразный стул, легкое вздутие живота, высокая лихорадка. Прогноз: средняя частота первых ремиссий при остром миелобластном и миеломонобластном лейкозах при современной терапии достигает 60%. Продолжительность первой ремиссии превышает 2 года. Онкологическое выздоровление наблюдается у 10% больных всех возрастов.
Редкие формы острых лейкозов
Макрофагальный острый лейкоз - важнейший и постоянный его клинический признак - высокая асептическая лихорадка, иногда с ознобами. Часто наблюдается гепатоспленомегалия. Бластная инфильтрация печени часто осложняется желтухой.
Мегакариобластный острый лейкоз - клиническая картина сходна с другими острыми лейкозами, в крови и костном мозге наряду с недифференцируемыми бластными клетками присутствуют и мегакариобласты.
Миелодиспластические синдромы (МДС) — гетерогенная группа гемопатий, куда входят разнообразные заболевания неодинаковой природы, сопровождающиеся как значительными изменениями костного мозга, так и его функциональными нарушениями, часто трансформирующиеся в высокобластные острые лейкозы. Разнообразные гемопатии, объединенные экспертами ВОЗ в группу под общим названием МДС, имеют общую причину - повреждение клеток на уровне, близком к стволовой кроветворной клетке. В отличие от других гемобластозов, при МДС клеточный апоптоз сохранен или усилен, что и формирует периферическую би/пан/цитопению на фоне гиперплазии костного мозга.
Преобладающим симптомом является малокровие. Часть гемопатий. входящих в этот «сборник», иногда называют предлейкозом. Современные данные показывают, что предлейкоз - это доклиническая стадия острого лейкоза, при которой ограниченное количество бластов еще не вызывает явных клинических расстройств. Гематологическими признаками формирующегося острого лейкоза являются: нарастающая анемия без ретикулоцитоза; «необъяснимые» гранулоцитопении; «беспричинный» моноцитоз без явной гиперплазии костного мозга в трепанате; хронический нерезко выраженный аутоиммунный гемолиз, немногочисленные очаги гиперплазии в трепанате костного мозга (через несколько лет возможна трансформация в острый лейкоз). Варианты МДС отмечаются в основном у лиц пожилого возраста (старше 60 лет). Характерна слабость, потливость, утомляемость, одышка. Умеренное увеличение печени и селезенки наблюдается только при варианте ХММЛ: лимфоузлы как правило в норме. Лейкозы, развившиеся из миелодиспластических синдромов, отличаются меньшей чувствительностью к терапии, чем первичные острые нелимфобластные лейкозы.
Монобластный острый лейкоз наблюдается весьма редко. Клиническая картина этой формы лейкоза напоминает острый миелобластный лейкоз, однако, в этой форме значительнее выражены интоксикация и гипертермия, чаще наблюдаются некротические изменения слизистой оболочки рта и глотки. Весьма характерна лейкемическая инфильтрация десен, ведущая к гипертрофии сосочков («лейкемический гингивит»). Нередко развивается инфильтрация миндалин и десен, а на поздних этапах прогрессии возникают опухолевые пролифераты во всех внутренних органах, лейкемиды кожи и т.д.
Плазмобластный острый лейкоз отмечается во всех возрастах, в т.ч. у молодых лиц и у детей, характерно острое клиническое течение. Наблюдается увеличение лимфоузлов, селезенки, печени. В сыворотке крови выявляется М-градиент - моноклональный иммуноглобулин (парапротеин).
Промиелоцитарный острый лейкоз - довольно редко встречается - не более 7% всех острых лейкозов. Прежде это заболевание отличалось тяжелейшим прогнозом (массивная кровопотеря или геморрагический инсульт уже в первые недели от момента диагностики), а теперь - это одна из наиболее курабельных форм, при которой наблюдается «онкологическое» излечение на фоне дозревания лейкозных бластов до нормальных форм.
Эритромиелоз острый, эритробластный острый лейкоз, болезнь Ди Гульельмо - редкая форма острого лейкоза. В анамнезе больных острым эритромиелозом часто встречается лучевая или химиотерапия; нередко выявляется как вторичный лейкоз у лиц, пролеченных по поводу лимфогранулематоза, миеломной болезни, эритремии.
Генетические маркеры
Транслокации после крупных делеций и дупликаций являются самыми частыми хромосомными аномалиями. Различают два основных типа специфических хромосомных транслокаций:
- первый приводит к активации протоонкогенов без их структурных изменений;
- в результате второго из фрагментов двух ранее существовавших генов образуются совершенно новые гены, так называемые химерные.
Транслокации хромосомы 8 с хромосомами 2, 14 и 22, в результате которых происходит перемещение протоонкогена MYC (8q24) в область генов иммуноглобулинов IGH (14q32), IGK (2p12) и IGL (22q11), – классический пример аномалии первого типа. Эти гены обладают очень сильными регулярными элементами, которые начинают постоянно активировать MYC. Как правило, не участвует в перестройке первый некодирующий экзон протоонкогена. В 90% случаев лимфомы Беркитта выявляются именно эти транслокации.
В результате транслокаций, инверсий и небольших делеций достаточно часто появляются химерные гены с новой агрессивной функцией белкового продукта – слитных белков. В 1973 г описана специфическая транслокация хромосом 9 и 22, приводящая к появлению филадельфийской хромосомы при хронической миелоидной лейкемии, – классический пример транслокаций второго типа. В результате появляется ген, кодирующий слитный белок, имеющий свойства тирозинкиназы, подобной ABL, но с аномальными трансформирующими свойствами.
Описано 358 химерных, формируемых 337 различными генами. Химерные гены определены практически во всех типах злокачественных опухолей (Mitelman et al., 2007). В злокачественных опухолях кроветворной системы встречается почти 75% химерных генов. При остром миелоидном лейкозе описано 267 химерных генов, при остром лимфобластном лейкозе – 155, в солидных опухолях – только 75. Для каждого типа заболевания характерны определённые перестройки, им соответствует определенная клиническая картина. Зачастую транслокации позволяют поставить точный диагноз, подобрать лечение и прогнозировать течение заболевания.
Химерные гены могут провоцировать развитие опухоли определённого гистогенетического типа. Образующийся в результате транслокации t(12;15)(p13;q25) химерный ген ETV6-NTRK3 вызывает ОМЛ, мезобластную нефрому, фибросаркому, аденокарциному молочной железы.
Молекулярная цитогенетика, в частности, многоцветная флуоресцентная гибридизация in situ, SKY и сравнительная геномная гибридизация позволяют осуществлять поиск новых химерных генов. Методы ОТ-ПЦР (обратная транскрипция мРНК-ПЦР) или прямого секвенирования позволяют диагностировать химерные гены, характерные для определённой опухоли.


Согласен Данный веб-сайт содержит информацию для специалистов в области медицины. В соответствии с действующим законодательством доступ к такой информации может быть предоставлен только медицинским и фармацевтическим работникам. Нажимая «Согласен», вы подтверждаете, что являетесь медицинским или фармацевтическим работником и берете на себя ответственность за последствия, вызванные возможным нарушением указанного ограничения. Информация на данном сайте не должна использоваться пациентами для самостоятельной диагностики и лечения и не может быть заменой очной консультации врача.
Лимфогранулематоз (лимфома Ходжкина)

«Лимфома Ходжкина — одно из первых онкозаболеваний, которое было успешно пролечено для большой группы больных во всем мире. Благодаря современной системе TomoTherapy®, установленной в нашей клинике, мы лечим даже самые тяжелые формы лимфомы Ходжкина со сложными локализациями. Облучение на нашем аппарате ведется одновременно для нескольких зон и нам удается выдерживать индивидуальную дозу для каждой из них».
Что такое лимфогранулематоз?
Лимфома Ходжкина (синонимы: лимфогрануломатоз, болезнь Ходжкина) — это опухолевое заболевание лимфатической системы. Патология составляет примерно 1% всех онкологических заболеваний, ежегодно регистрируемых в мире. Лимфогрануломатоз может проявиться в любом возрасте, однако отмечается два пика заболевания: первый приходится на возраст 20-29 лет, второй на 60 лет и старше.
Лимфогранулематоз — это заболевание, которое, как уже говорилось, поражает лимфатическую систему человека. Онколог может полностью подтвердить диагноз, если в новообразованиях будут присутствовать так называемые гигантские клетки Березовского-Штернберга-Рида, которые “сообщают” врачу об аномалии в нормальном функционировании тканей.
Болезнь Ходжкина — злокачественная опухоль, но рак это или нет? С медицинской точки зрения, данное новообразование не входит в группу раковых патологий, так как системность поражения не соответствует и терапия болезни достаточно простая. Мужчины подвержены развитию патологии больше, чем женщины. Опухолевые клетки Березовского-Штернберга-Рида могут обнаружить только специалисты в результате обследования пациента после сдачи биохимического анализа крови. Именно поэтому настолько важно обратиться в хороший медицинский центр.
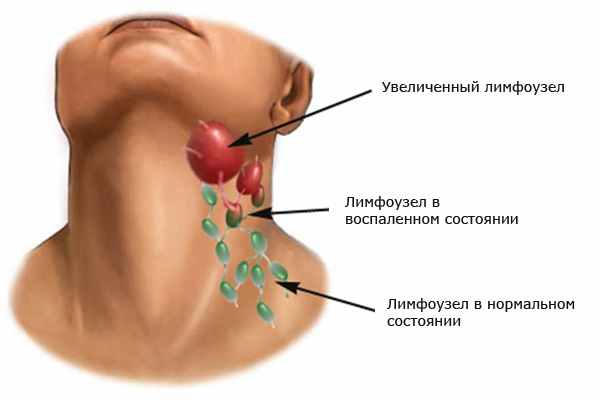
Лимфогранулематоз — причины
Причины развития лимфомы до конца еще не выявлены. Установлено, что пациенты с иммунодефицитными состояниями гораздо чаще подвергаются заболеванию. В 40-60% случаев болезни Ходжкина у людей обнаруживается вирус Эпштейна-Барра. Основываясь на данном факте, рассматривается определенная роль вируса в развитии лимфомы.
Лимфомы — это не наследственное заболевание, заражение от человека к человеку не возможно. Также редко причиной лимфомы могут быть врожденные генетические дефекты. Этиология достоверно не понятна, но в медицине есть данные о повышенной заболеваемости среди жителей определенных регионов, что дает возможность сделать выводы о влиянии вирусов и внешних факторов на частоту заболеваемости среди населения.
Среди предрасполагающих факторов медики выделили следующие:
- Наследственность (если у кого-то из родственников были диагностированы патологии лимфоидной ткани, то велика вероятность развития данной патологии).
- Иммунодефицит с рождения или приобретенный в течение жизни.
- Аутоиммунные патологии, например, красная волчанка.
Отмечается тесная связь инфекционного мононуклеоза с развитием болезни Ходжкина. Тем не менее, эта патология развивается не у всех людей, у которых присутствует вирус в крови. Согласно проводимым исследованиям изолированное воздействие этого фактора определяет вероятность развития патологии только лишь в 0,1%. При этом вирус Эпштейна-Барра по статистике выявляется у 90 процентов населения планеты. В группу риска входят также люди с разного рода иммунодефицитными состояниями, например те, у кого диагностирована ВИЧ-инфекция.
Симптомы лимфогранулематоза
Симптомы патологии проявляются по-разному: заболевание начинает свое развитие в лимфоузлах, а дальнейший болезненный процесс может переходить практически на все органы, зачастую сопровождаясь различно выраженными интоксикационными проявлениями. Картину лимфогранулематоза определяет локализация опухоли, поражение того или иного органа.
Первый признак патологии — увеличение лимфоузлов. В 60-75% случаев онкопроцесс начинается в шейно-надключичных лимфоузлах, чаще поражается правая сторона. Обычно увеличение лимфоузлов не приводит к ухудшению самочувствия и человек не замечает никаких изменений. В 15-20% случаев изначально поражаются лимфоузлы средостения. Это может быть случайно обнаружено при рентгенологическом обследовании или когда увеличение лимфоузлов становится значительным. Во втором случае больные обращаются в лечебное учреждение с жалобами на кашель, одышку, отечность и синюшность лица, иногда — боли за грудиной.
В единичных случаях лимфогранулематоз начинается с поражения парааортальных лимфоузлов, у больного могут появиться боли в спине, усиливающиеся ночью. У 5-10% больных лимфогранулематоз начинается с лихорадки, похудания, ночных потов, а кожный зуд встречается у 20-25% больных. При дальнейшем развитии лимфогранулематоза возможно поражение всех лимфоидных и остальных систем, органов организма. Наиболее часто кроме лимфатических узлов лимфома Ходжкина поражает кости и легочную ткань. Вовлечение в процесс легких клинически может никак не проявляться, а вот при повреждении костей появляются боли. Наряду с локальной симптоматикой пациента серьезно мучают общие проявления (симптомы группы B):
- Лихорадочное состояние (высокая температура тела), которое не проходит более 7 дней.
- Сильнейшее отделение пота в период ночного сна.
- Не контролируемая потеря массы тела (более 10 процентов потери веса в течение полугода).
Симптомы «B» характеризуют наиболее серьезное протекание патологии и дают возможность определить надобность назначения интенсивного лечения. Среди прочей симптоматологии характерными для данного заболевания можно выделить:
- Состояние слабости, нет желания принимать какую-либо пищу, полный упадок сил.
- Кожные покровы сильно чешутся.
- Кашель, болевой синдром в грудной клетке, человеку трудно дышать.
- Болевой синдром в области брюшины, расстройство пищеварительной системы.
- Асцит.
- Болевой синдром в костной ткани.
- Увеличение лимфатических узлов.
В ряде случаев первые симптомы опасной патологии в течение долгого срока проявляются постоянным ощущением вялости и быстрой усталостью. Проблемы с дыханием развиваются, когда идет увеличение лимфоузлов внутри грудины. По мере разрастания узлов и продолжения поражения организма они начинают давить на трахею, провоцируют развитие кашля и прочих проблем с дыхательной системой. Эти симптомы становятся более ярко выраженными, когда человек лежит. Иногда человек может ощущать болевой синдром за грудой клеткой.
Важно понимать: как только развились первые симптомы лимфомы Ходжкина (желательно до того, как увеличились лимфатические узлы и пошло поражение внутренних органов), нужно посетить хороший медицинский центр, где врач проведет комплексную диагностику и подберет эффективный метод лечения.
Читайте также: