Кардиомиопатии. Эпидемиология или распространенность кардиомиопатий.
Добавил пользователь Валентин П. Обновлено: 29.01.2026
В статье представлен аналитический обзор современных источников научной литературы, освещающих вопросы эпидемиологии, патогенеза и акушерской тактики при перипартальной кардиомиопатии (ПКМП). Данные о заболеваемости ПКМП крайне гетерогенны и зависят от географического региона. Смертность при данной патологии достигает 4-28%. К факторам риска развития ПКМП относятся многоплодная беременность и рождение нескольких детей, семейный анамнез, курение, сахарный диабет, артериальная гипертензия, преэклампсия, пониженное питание, старший или подростковый возраст матери и длительное лечение агонистами бета-адренорецепторов. В генезе заболевания ведущая роль отводится генетическому фактору, что подтверждается семейным анамнезом и изменением распространенности заболевания в зависимости от географического расположения. Описана патогенетическая роль вазоингибина - изоформы пролактина, обладающей мощным антиангиогенным, проапоптотическим, провоспалительным и вазоконстрикторным действием. Приведены данные, свидетельствующие о значимости нарушения соотношения ангио- и антиангиогенных факторов. В настоящее время отсутствуют общепринятые клинические рекомендации для акушеров-гинекологов по ведению беременных, рожениц и родильниц с ПКМП. Это обусловлено малым количеством накопленных данных о заболевании. При родоразрешении экстренное кесарево сечение показано в случае наличия у беременной острой сердечной недостаточности и потребности в инотропных лекарственных средствах и/или инвазивной терапии. В то же время, по мнению экспертов Европейского общества кардиологов (ESC, 2018), при отсутствии описанных выше состояний план родоразрешения может быть пересмотрен в сторону естественных родов.
Ключевые слова
Об авторах
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Рудаева Елена Владимирова - кандидат медицинских наук, доцент кафедры акушерства и гинекологии им. Г.А. Ушаковой.
ул. Ворошилова, 22а, Кемерово, 650056.
Е.В. Рудаева заявляет об отсутствии конфликта интересов.
Хмелева Ирина Анатольевна - заведующая отделением кардиологии.
пр. Октябрьский, 22, Кемерово, 650066.
И.А. Хмелева заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Мозес Кира Борисовна - ассистент кафедры поликлинической терапии и сестринского дела.
ул. Ворошилова, 22а, Кемерово, 650056.
К.Б. Мозес заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Мозес Вадим Гельевич - доктор медицинских наук, профессор кафедры акушерства и гинекологии им. Г.А. Ушаковой.
ул. Ворошилова, 22а, Кемерово, 650056.
В.Г. Мозес заявляет об отсутствии конфликта интересов.
Захаров Игорь Сергеевич - доктор медицинских наук, заместитель главного врача государственного.
пр. Октябрьский, 53/1, Кемерово, 650066.
И.С. Захаров заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Елгина Светлана Ивановна - доктор медицинских наук, профессор кафедры акушерства и гинекологии им. Г.А. Ушаковой.
ул. Ворошилова, 22а, Кемерово, 650056.
С.И. Елгина заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Марцияш Алексей Алексеевич - доктор медицинских наук, профессор кафедры неврологии, нейрохирургии, медицинской генетики и медицинской реабилитации.
ул. Ворошилова, 22а, Кемерово, 650056.
А.А. Марцияш заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации; Клинический консультативно-диагностический центр
Россия
Колпинский Глеб Иванович - доктор медицинских наук, профессор кафедры лучевой диагностики, лучевой терапии КемГМУ МЗ РФ; главный врач государственного, Клинический консультативно-диагностический центр.
ул. Ворошилова, 22а, Кемерово, 650056; пр. Октябрьский, 53/1, Кемерово, 650066.
Г.И. Колпинский заявляет об отсутствии конфликта интересов.
Кемеровский государственный медицинский университет Министерства здравоохранения Российской Федерации
Россия
Шапкин Александр Анатольевич - кандидат медицинских наук, доцент кафедры факультетской хирургии.
ул. Ворошилова, 22а, Кемерово, 650056.
А.А. Шапкин заявляет об отсутствии конфликта интересов.
Список литературы
1. Pfeffer T.J., Hilfiker-Kleiner D. Pregnancy and heart disease: pregnancy-associated hypertension and peripartum cardiomyopathy. Curr Probl Cardiol. 2018; 43 (9): 364-388. doi:10.1016/j.cpcardiol.2017.10.005
2. Vatutin N.T., Taradin G.G., Popelnukhina L.G., Gritzenko Y.P., Sidorenko I.A. Treatment of peripartum cardiomyopathy (review). Archive of internal medicine. 2017; 7 (5): 340-349. doi:10.20514/2226-6704-2017-7-5-340-349
3. Kamiya C.A., Yoshimatsu J., Ikeda T. Peripartum cardiomyopathy from a genetic perspective. Circ J. 2016; 80 (8): 1684-8. doi: 10.1253/circj.CJ-16-0342
4. Lewey J., Levine L., Elovitz M., Irizarry O.C., Arany Z. Importance of early diagnosis in peripartum cardiomyopathy. Hypertension. 2020; 75 (1): 91-97. doi:10.1161/hypertensionaha.119.13291
6. Masoomi R., Shah Z., Arany Z., Gupta K. Peripartum cardiomyopathy: an epidemiologic study of early and late presentations. Pregnancy Hypertens. 2018; 13: 273-278. doi:10.1016/j.preghy.2018.06.018
7. Hakata S., Umegaki T., Soeda T., Nishimoto K., Ando A., Anada N., Uba T., Sumi C., Kamibayashi T. Bromocriptine use for sudden peripartum cardiomyopathy in a patient with preeclampsia: a case report. JA Clin Rep. 2019; 5 (1): 38. doi:10.1186/s40981-019-0256-8
8. Elkayam U. Clinical characteristics of peripartum cardiomyopathy in the United States: diagnosis, prognosis, and management. J Am Coll Cardiol 2011; 58: 659-670.
9. Kamiya C.A., Kitakaze M., Ishibashi-Ueda H., Nakatani S., Murohara T., Tomoike H., Ikeda T. Different characteristics of peripartum cardiomyopathy between patients complicated with and without hypertensive disorders. Results from the Japanese Nationwide survey of peripartum cardiomyopathy. Circ J 2011; 75: 1975-1981.
10. Karaye K.M., Ishaq N.A., Sa'idu H. Balarabe S.A., Talle M.A., Isa M.S., Adamu U.G., Umar H., Okolie H.I., Shehu M.N., Mohammed I.Y., Sanni B., Ogah O.S., Oboirien I., Umuerri E.M., Mankwe A.C., Shidali V.Y., Njoku P., Dodiyi-Manuel S., Shogade T.T., Olunuga T., Ojji D., Josephs V., Mbakwem A.C., Tukur J., Isezuo S.A.; PEACE Registry Investigators. Incidence, clinical characteristics, and risk factors of peripartum cardiomyopathy in nigeria: Results from the PEACE registry. ESC Heart Fail. 2020; 7 (1): 235-243. doi: 10.1002/ehf2.12562
11. Kolte D., Khera S., Aronow W.S., Palaniswamy C., Mujib M., Ahn C., Jain D., Gass A., Ahmed A., Panza J.A., Fonarow G.C. Temporal trends in incidence and outcomes of peripartum cardiomyopathy in the United States: a nationwide population-based study. J Am Heart Assoc. 2014; 3: e001056. doi: 10.1161/JAHA.114.001056.
12. Thompson J.L., Kuklina E.V., Bateman B.T., Callaghan W.M., James A.H., Grotegut C.A. Medical and obstetric outcomes among pregnant women with congenital heart disease. Obstet Gynecol. 2015; 126 (2): 346-54. doi:10.1097/aog.0000000000000973
13. Grotegut C.A., Kuklina E.V., Anstrom K.J. Factors associated with the change in prevalence of cardiomyopathy at delivery in the period 2000-2009: A population-based prevalence study. BJOG. 2014; 121 (11): 1386-94. doi: 10.1111/1471-0528.12726
16. Мозес В.Г. Роль системного поражения соединительной ткани в генезе варикозного расширения вен малого таза у подростков. Казанский медицинский журнал. 2006; 87(2): 102-104
17. Рудаева Е.В., Мозес В.Г., Кашталап В.В., Захаров И.С., Елгина С.И., Рудаева Е.Г. Врожденные пороки сердца и беременность. Фундаментальная и клиническая медицина. 2019; 4 (3): 102-112
18. Safirstein J.G., Ro A.S., Grandhi S., Wang L., Fett J.D., Staniloae C. Predictors of left ventricular recovery in a cohort of peripartum cardiomyopathy patients recruited via the internet. Int J Cardiol. 2012; 154: 27-31. doi: 10.1016/j.ijcard.2010.08.065.
19. McNamara D.M., Elkayam U., Alharethi R., Damp J., Hsich E., Ewald G. et al. Clinical outcomes for peripartum cardiomyopathy in North America: results of the IPAC Study (Investigations of Pregnancy-Associated Cardiomyopathy). J Am Coll Cardiol. 2015; 66: 905-14. doi: 10.1016/j.jacc.2015.06.1309
20. Brar S.S., Khan S.S., Sandhu G.K., Jorgensen M.B., Parikh N., Hsu J.W., Shen A.Y. Incidence, mortality, and racial differences in peripartum cardiomyopathy. Am J Cardiol 2007; 100: 302-304. doi: 10.1016/j.amjcard.2007.02.092.
21. Lee Y.Z.J., Judge D.P. The role of genetics in peripartum cardiomyopathy. Review J Cardiovasc Transl Res. 2017; 10 (56): 437-445. doi: 10.1007/s12265-017-9764-y
22. McNally E.M., Puckelwartz M.J. Genetic variation in cardiomyopathy and cardiovascular disorders. Circ J 2015; 79: 1409-1415. doi: 10.1253/circj.CJ-15-0536.
23. Morales A., Painter T., Li R., iegfried J,D,, Li D,, Norton N,, Hershberger R,E. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. 2010; 121 (20): 2176-82. doi:10.1161/circulationaha.109.931220
24. van Spaendonck-Zwarts K.Y., van Tintelen J.P., van Veldhuisen D.J., van der Werf R., Jongbloed J.D., Paulus W.J., Dooijes D., van den Berg M.P. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010; 121 (20): 2169-75. doi: 10.1161/circulationaha.109.929646
25. Шибельгут Н.М., Захаров И.С., Мозес В.Г. Клинико-биохимические проявления недифференцированных форм дисплазии соединительной ткани у беременных с варикозной болезнью вен малого таза. Саратовский научно-медицинский журнал. 2010; 6 (1). С. 56-60
27. van Spaendonck-Zwarts K.Y., van Tintelen J.P., van Veldhuisen D.J., van der Werf R., Jongbloed J.D., Paulus W.J., Dooijes D., van den Berg M.P. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010; 121: 2169-2175. doi: 10.1161/CIRCULATIONAHA.109.929646.
28. van Spaendonck-Zwarts K.Y., Posafalvi A., van den Berg M.P., Hilfiker-Kleiner D., Bollen I.A., Sliwa K., Alders M., Almomani R., van Langen I.M., van der Meer P., Sinke R.J., van der Velden J., Van Veldhuisen D.J., van Tintelen J.P., Jongbloed J.D. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J 2014; 35: 2165-2173. doi: 10.1093/eurheartj/ehu050.
30. Onusko E., McDermott M.R., Robbins N. Probenecid treatment improves outcomes in a novel mouse model of peripartum cardiomyopathy. PLoS One. 2020; 15 (3): e0230386. doi: 10.1371/journal.pone.0230386
31. de la Torre P., Perez-Lorenzo M.J., Alcazar-Garrido A., Flores A.I. Cell-based nanoparticles delivery systems for targeted cancer therapy: lessons from anti-angiogenesis treatments. Molecules. 2020; 25 (3): 715. doi: 10.3390/molecules25030715
32. Cunningham F.G., Byrne J.J., Nelson D.B. Peripartum cardiomyopathy. Obstet Gynecol. 2019; 133 (1): 167-179. doi: 10.1097/AOG.0000000000003011
33. Yang S.H., Sharrocks A.D., Whitmarsh A.J. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013; 513 (1): 1-13. doi:10.1016/j.gene.2012.10.033
35. Hilfiker-Kleiner D., Kaminski K., Podewski E., Bonda T., Schaefer A., Sliwa K. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007; 128: 589-600. doi: 10.1016/j.cell.2006.12.036.
36. Ricke-Hoch M., Bultmann I., Stapel B., Condorelli G., Rinas U., Sliwa K., Scherr M., Hilfiker-Kleiner D. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovasc Res. 2014; 101(4): 587-96. doi: 10.1093/cvr/cvu010
37. Halkein J., Tabruyn S.P., Ricke-Hoch M., Haghikia A., Nguyen N.Q., Scherr M., Castermans K., Malvaux L., Lambert V., Thiry M., Sliwa K., Noel A., Martial J.A., Hilfiker-Kleiner D., Struman I. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013; 123: 2143-54. doi: 10.1172/JCI64365.
38. Sliwa K., Blauwet L., Tibazarwa K., Libhaber E., Smedema J.P., Becker A., McMurray J., Yamac H., Labidi S., Struman I., Hilfiker-Kleiner D. Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: a proof-of-concept pilot study. Circulation. 2010; 121: 14651473. doi: 10.1161/CIRCULATIONAHA.109.901496.
39. Hilfiker-Kleiner D., Meyer G.P., Schieffer E., Goldmann B., Podewski E., Struman I., Fischer P., Drexler H. Recovery from postpartum cardiomyopathy in 2 patients by blocking prolactin release with bromocriptine. J. Am. Coll. Cardiol. 2007; 50: 2354-2355. doi: 10.1016/j.jacc.2007.10.006.
41. Koenig T., Bauersachs J., Hilfiker-Kleiner D. Bromocriptine for the treatment of peripartum cardiomyopathy. Card Fail Rev. 2018; 4 (1): 46-49. doi: 10.15420/cfr.2018:2:2
42. Avila M.S., Siqueira S.R.R., Ferreira S.M.A., Bocchi E.A. Prevention and treatment of chemotherapy-induced cardiotoxicity. Methodist Debakey Cardiovasc J. 2019; 15 (4): 267-273. doi:10.14797/mdcj-15-4-267
43. Behrens I., Basit S., Lykke J.A., Ranthe M.F., Wohlfahrt J., Bundgaard H., Melbye M., Boyd H.A. Hypertensive disorders of pregnancy and peripartum cardiomyopathy: a nationwide cohort study. PLoS One. 2019; 14 (2): e0211857. doi: 10.1371/journal.pone.0211857
44. Bello N., Rendon I.S.H., Arany Z. The relationship between pre-eclampsia and peripartum cardiomyopathy: a systematic review and meta-analysis. Review J Am Coll Cardiol. 2013; 62 (18): 1715-1723. doi: 10.1016/j.jacc.2013.08.717
47. Rajakumar A., Michael H.M., Rajakumar P.A., Shibata E., Hubel C.A., Karumanchi S.A., Thadhani R., Wolf M., Harger G., Markovic N. Extra-placental expression of vascular endothelial growth factor receptor-1, (Flt-1) and soluble Flt-1 (sFlt-1), by peripheral blood mononuclear cells (PBMCs) in normotensive and preeclamptic pregnant women. Placenta. 2005; 26: 563-73. doi: 10.1016/j.placenta.2004.09.001.
48. Patten I.S., Rana S., Shahul S., Rowe G.C., Jang C., Liu L., Hacker M.R., Rhee J.S., Mitchell J., Mahmood F., Hess P., Farrell C., Koulisis N., Khankin E.V., Burke S.D., Tudorache I., Bauersachs J., del Monte F., Hilfiker-Kleiner D., Karumanchi S.A., Arany Z. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012; 485 (7398): 333-338. doi: 10.1038/nature11040
49. van Opbergen C.J.M., den Braven L., Delmar M., van Veen T.A.B. Mitochondrial dysfunction as substrate for arrhythmogenic cardiomyopathy: a search for new disease mechanisms. Front Physiol. 2019; 10: 1496. doi: 10.3389/fphys.2019.01496
50. Bello N.A., Zoltan Arany Z. Molecular mechanisms of peripartum cardiomyopathy: A vascular/hormonal hypothesis. Trends Cardiovasc Med. 2015; 25 (6): 499-504. doi: 10.1016/j.tcm.2015.01.004
52. ESC Guidelines on the Management of Cardiovascular Diseases During Pregnancy: The Task Force on the Management of Cardiovascular Diseases During Pregnancy of the European Society of Cardiology (ESC). Practice Guideline Eur Heart J. 2011; 32 (24): 3147-97. doi: 10.1093/eurheartj/ehr218
53. Regitz-Zagrosek V., Roos-Hesselink J.W., Bauersachs J., Blomstrom-Lundqvist C., Ci'lkova R., De Bonis M., Iung B., Johnson M. 2018 ESC Guidelines for the Management of Cardiovascular Diseases During Pregnancy. Kardiol Pol. 2019; 77 (3): 245-326. doi: 10.5603/KP.2019.0049
55. Edupuganti M.M., Ganga V. Acute myocardial infarction in pregnancy: Current diagnosis and management approaches. Indian Heart J. 2019; 71 (5): 367-374. doi: 10.1016/j.ihj.2019.12.003
56. Marstrand P., Picard K., Lakdawala N.K. Second hits in dilated cardiomyopathy. Curr Cardiol Rep. 2020; 22 (2): 8. doi: 10.1007/s11886-020-1260-3
57. Seeland U., Goldin-Lang P., Regitz-Zagrosek V. Cardiovascular diseases in pregnancy: facts of the new guideline. Dtsch Med Wochenschr. 2012; 137 (31-32): 156872. doi: 10.1055/s-0032-1305187
59. Arany Z., Feldman A.M. To breastfeed or not to breastfeed with peripartum cardiomyopathy. JACC Basic Transl Sci. 2019; 4 (3): 301-303. doi: 10.1016/j.jacbts.2019.03.005
60. Koczo A., Marino A., Jeyabalan A., Elkayam U., Cooper L.T., Fett J., Briller J., Hsich E., Blauwet L., McTiernan C., Morel P.A., Hanley-Yanez K., McNamara D.M.; IPAC Investigators. Breastfeeding, cellular immune activation, and myocardial recovery in peripartum cardiomyopathy. JACC Basic Transl Sci. 2019 Jun 24; 4 (3): 291-300. doi: 10.1016/j.jacbts.2019.01.010
Кардиомиопатии. Эпидемиология или распространенность кардиомиопатий.
Кардиомиопатии. Эпидемиология или распространенность кардиомиопатий.
Определение кардиомиопатии (КМП) в настоящее время носит самый общий характер — это заболевание миокарда, приводящее к дисфункции сердца. Однако характерной чертой для всех вариантов болезни является формирование структурных изменений в миокарде и развитие недостаточности кровообращения. Попытки систематизировать данные аномалии основаны на преобладающих патофизиологических, этиологических или патогенетических факторах.
В соответствии с классификацией 1995 г. выделяют четыре основные группы патологий: дилатационную кардиомиопатию, гипертрофическую кардиомиопатию, рестриктивную КМП и аритмогенную правожелудочковую КМП. Примерно у 40% пациентов удается установить этиологию заболевания. В частности, первичные «идиопатические» варианты кардиомиопатии (дилатационные, гипертрофические, рестриктивные) либо представляют собой генетически обусловленные семейные случаи заболевания, либо являются следствием впервые возникших генных мутаций. Результатом таких мутаций является нарушение нормальной структуры сократительных белков миокарда с последующим снижением насосной функции сердца и развитием дилатации полостей.

Отдельный раздел составляют так называемые неклассифицированные кардиомиопатии, к которым относят некомпактный миокард, фиброэластоз, митохондриальные заболевания и др. Кроме того, существуют специфические кардиомиопатии - патология миокарда, сочетающаяся с другими сердечными или системными заболеваниями. Основанием для диагноза кардиомиопатия в этих случаях является несоответствие тяжести нарушения функции сердца и степени первичного заболевания (коронарной или клапанной патологии, артериальной гипертензии). Сюда же относят поражения сердца метаболического происхождения (эндокринные, связанные с дефицитом некоторых субстанций, патологическими инфильтрациями миокарда, амилоидной болезнью), сопряженные с системными заболеваниями соединительной ткани, мышечными дистрофиями, аллергическими и токсическими реакциями, и некоторые другие.
Точная частота заболеваний неизвестна, так как во многих случаях они протекают бессимптомно. Выявляемость первичных КМП в детском возрасте колеблется в широких пределах. Так, в Финляндии на 100 000 детей она составляет 0,65, в США — 1,13, в Австралии — 1,24. Основное количество (более 50%) случаев представлено дилатационной кардиомиопатией (ДКМП), около 40% - гипертрофической кардиомиопатией (ГКМП) и остальные — неспецифическими кардиопатиями. Частота заболевания у грудных детей почти в 12 раз выше, чем в остальных возрастных группах. Среди плодов с патологией сердца КМП составляют 8,9% случаев, среди новорожденных — 3%.
Естественное течение кардиомиопатий. Кардиомиопатии представляют серьезную проблему у детей: около 40% из них умирают или требуют трансплантации сердца в течение двух лет после появления клинических симптомов. При выявлении патологии во внутриутробном периоде около 13% беременностей прерывают, 63% плодов погибает в перинатальном периоде.
Наиболее часто встречающиеся кардиомиопатии у новорожденных представлены ниже.
- Вернуться в оглавление раздела "Кардиология."
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Кардиомиопатии. Эпидемиология или распространенность кардиомиопатий.
Первый МГМУ им. И.М. Сеченова;
Московский НИИ педиатрии и детской хирургии
Отделение кардиохирургии клиники аортальной и сердечно-сосудистой хирургии Первого МГМУ им. И.М. Сеченова
Кафедра сердечно-сосудистой хирургии и инвазивной кардиологии Первого МГМУ им. И.М. Сеченова (зав. — д.м.н. Р.Н. Комаров), Российский научный центр хирургии им. акад. Б.В. Петровского (дир. — акад. РАН Ю.В. Белов), Москва, Россия
кафедра сердечно-сосудистой хирургии и инвазивной кардиологии Первого Московского медицинского университета им. И.М. Сеченова, Москва, Россия
ФГАОУ ВО «ПМГМУ им. И.М. Сеченова», Москва, Россия
ФГАОУ ВО «ПМГМУ им. И.М. Сеченова», Москва, Россия
РНЦХ им. акад. Б.В. Петровского РАМН
Современный взгляд на диагностику и лечение гипертрофической кардиомиопатии
Журнал: Кардиология и сердечно-сосудистая хирургия. 2019;12(1): 38‑44
Первый МГМУ им. И.М. Сеченова;
Московский НИИ педиатрии и детской хирургии
Гипертрофическая кардиомиопатия является распространенным генетическим заболеванием с частотой встречаемости около 0,2%. Естественное течение заболевания может приводить к возникновению и прогрессированию сердечной недостаточности, внезапной сердечной смерти. В статье рассматривается исторический аспект, описаны современные методы диагностики, в том числе молекулярно-генетический, дана интерпретация актуальным методам консервативного и хирургического лечения этой патологии. Особое внимание уделено перспективным направлениям развития методов диагностики и лечения заболевания.
Первый МГМУ им. И.М. Сеченова;
Московский НИИ педиатрии и детской хирургии
Отделение кардиохирургии клиники аортальной и сердечно-сосудистой хирургии Первого МГМУ им. И.М. Сеченова
Кафедра сердечно-сосудистой хирургии и инвазивной кардиологии Первого МГМУ им. И.М. Сеченова (зав. — д.м.н. Р.Н. Комаров), Российский научный центр хирургии им. акад. Б.В. Петровского (дир. — акад. РАН Ю.В. Белов), Москва, Россия
кафедра сердечно-сосудистой хирургии и инвазивной кардиологии Первого Московского медицинского университета им. И.М. Сеченова, Москва, Россия
ФГАОУ ВО «ПМГМУ им. И.М. Сеченова», Москва, Россия
ФГАОУ ВО «ПМГМУ им. И.М. Сеченова», Москва, Россия
РНЦХ им. акад. Б.В. Петровского РАМН
Гипертрофическая кардиомиопатия (ГКМП) является распространенным генетическим заболеванием, причиной возникновения которого являются около 1400 мутаций в более чем 11 генах [1—9]. Согласно данным исследования CARDIA [10], частота ГКМП составляет 1:500, и естественное течение заболевания может приводить к сердечной недостаточности и внезапной сердечной смерти. ГКМП проявляется гипертрофией миокарда при отсутствии дилатации полости левого желудочка (ЛЖ), а также аномалиями хорд и папиллярных мышц. Выделяют группы пациентов с обструктивной и необструктивной формой ГКМП. Среди пациентов с обструктивной ГКМП наиболее часто (50—75% случаев) встречается обструкция выходного отдела ЛЖ, которая возникает на фоне переднего систолического движения (systolic anterior motion, SAM-синдром) передней створки митрального клапана и ее соприкосновением с гипертрофированной межжелудочковой перегородкой (МЖП) [11].
Хирургическое вмешательство является «золотым стандартом» лечения обструктивной ГКМП. В 1961 г. A. Morrow и E. Brockenbrough [13] описали алгоритм выполнения септальной миоэктомии (МЭ), однако выполнена данная операция была впервые еще в 1958 г. английским кардиохирургом W. Cleland [14].
В целом, вопрос эффективной диагностики и лечения ГКМП, а также проблема определения факторов, потенцирующих ее развитие, остаются открытыми. Данный вопрос требует комплексного подхода и дальнейшего изучения.
История
В 50-е годы ХХ века R. Brock, A. Morrow и E. Braunwald [15—17] сообщили о функциональной обструкции ЛЖ, которая имитировала аортальный стеноз. В последующем эта патология была названа «идиопатическим гипертрофическим субаортальным стенозом». Приблизительно в то же время английский патологоанатом D. Teare [18] отметил наличие асимметричной гипертрофии сердца с вовлечением в процесс МЖП и передней стенки ЛЖ у молодых пациентов. Зачастую симптоматически эта патология никак себя не проявляла, но иногда приводила к внезапной смерти. Морфологическая картина была одинаковой у всех пациентов и обусловливалась дезорганизацией пучков сердечной мышцы («миокардиальный беспорядок») с гипертрофией отдельных мышечных волокон и их ядер. Нередко морфологи и патологоанатомы обнаруживали участки фиброза в центре гипертрофированного миокарда. Генез этого проявления был определен, как постишемический. В то же время подозревали генетическую природу ГКМП, основанную на выявлении аутосомно-доминантного типа наследования данной патологии в семье у одного из погибших пациентов [19]. Крайне вариабельные морфологические проявления ГКМП приводили к трудностям в диагностике заболевания и, в итоге, к путанице в терминологии, которая зачастую основывалась на концепции подклапанного аортального стеноза или обструкции выходного отдела Л.Ж. Актуальный в настоящее время термин «гипертрофическая кардиомиопатия» был принят, когда выяснилось, что только у части пациентов патология проявляется обструкцией выходного отдела ЛЖ [20].
Новаторские исследования, проведенные в 60-е годы XX века, ознаменовали начало обширных клинических, морфологических, генетических и молекулярных исследований в этой области. Были получены знания, позволившие изучить процесс течения ГКМП и стратифицировать риски ее развития. В то время из-за отсутствия неинвазивных методов визуализации сердца применяли катетеризацию ЛЖ, что позволяло оценивать лишь такой гемодинамический показатель, как аорто-желудочковый градиент давления, однако данная методика не давала возможности определить причину его возникновения. Внедрение в конце 60-х годов эхокардиографии (ЭхоКГ) и широкое использование ультразвуковой кардиовизуализации помогли установить причину градиента давления между ЛЖ и аортой у больных с ГКМП, определить фенотипическую гетерогенность и патофизиологические последствия заболевания, установить истинную распространенность патологии. Исследование пациентов с помощью ЭхоКГ подтвердило эффективность хирургической миоэктомии и определило важную роль SAM-синдрома в патогенезе обструкции выходного отдела ЛЖ [21].
С эпидемиологической точки зрения, ЭхоКГ позволила оценить распространенность ГКМП в общей популяции и провести скрининг семей, в которых были отмечены случаи ГКМП [22].
Для диагностики ГКМП также применяли позитронно-эмиссионную и компьютерную томографию, которые позволяли определять перфузию миокарда и процесс ремоделирования коронарных сосудов [23—25].
С 1980 г. появился такой метод визуализации, как магнитно-резонансная томография (МРТ), которая в настоящее время является «золотым стандартом» диагностики ГКМП и позволяет оценить наличие фиброза миокарда и визуализировать различные фенотипические проявления ГКМП [26].
Инструментальная диагностика
Обструктивная ГКМП характеризуется гипертрофией миокарда ЛЖ при отсутствии дилатации полости желудочка, а также наличием обструкции его выходного тракта. Это приводит к возникновению и прогрессированию хронической сердечной недостаточности и повышает риск внезапной сердечной смерти. Основными причинами обструкции выходного отдела ЛЖ при ГКМП являются SAM-синдром и гипертрофия МЖП, а также аномальное расположение хорд и папиллярных мышц [27—33]. ЭхоКГ является одним из важнейших методов диагностики данной патологии и позволяет визуализировать причину обструкции выходного отдела ЛЖ, степень гипертрофии ЛЖ, а также с помощью допплеровского анализа оценить внутрижелудочковый и аорто-желудочковый градиенты давления [34—36]. Последний используют в качестве основного диагностического критерия для определения стратегии лечения заболевания. B. Maron [34] выделил 3 формы ГКМП:
1) обструктивная форма, при которой градиент давления в покое ≥ 30 мм рт.ст.;
2) латентная обструкция, при которой градиент давления составляет 30 мм рт.ст. в покое и ≥30 мм рт.ст. при нагрузке или провокационных пробах;
3) необструктивная форма, при которой градиент давления 30 мм рт. ст. как в покое, так и при применении провокационных проб или физической нагрузки [34].
Согласно M. Maron и соавт. [37], у пациентов с обструктивной формой ГКМП риск внезапной сердечной смерти гораздо выше, чем при необструктивной форме. Стоит отметить, что в 90% случаев обструктивной формы ГКМП выявляют утолщение МЖП до 13—15 мм [27]. Оценить толщину МЖП можно и с помощью чреспищеводной ЭхоКГ [38, 39]. При оценке переднего систолического движения передней створки митрального клапана обнаруживается неполная коаптация створок, что приводит к митральной регургитации [40].
С помощью допплеровского анализа можно получить достоверные данные о существующем градиенте давления между ЛЖ и аортой по пиковой скорости кровотока в выходном тракте ЛЖ [26, 41]. Однако стоит иметь в виду, что митральная регургитация может искажать данные о градиенте давления и приводить к гипердиагностике [42, 43].
МРТ сердца
Важным инструментальным методом диагностики является МРТ сердца. Данный метод имеет существенное преимущество перед традиционной ЭхоКГ, которое заключается в отсутствии ограничений доступа в виде узкого акустического окна [44]. МРТ позволяет более четко визуализировать митральный клапан, подклапанный аппарат и выводной отдел Л.Ж. Также МРТ оказалась более перспективной методикой в определении степени и распространенности гипертрофии ЛЖ, особенно, если гипертрофия имеет локальный характер — верхушка сердца, задняя часть МЖП.
Продольные и поперечные срезы выходного отдела ЛЖ позволяют в полной мере оценить переднее систолическое движение передней створки митрального клапана, установить турбулентный характер кровотока, а также точно рассчитать диаметр и площадь сечения выходного тракта Л.Ж. Площадь последнего ≤2,7 см 2 в систолу со 100% чувствительностью свидетельствует об обструктивной форме ГКМП [45]. Комбинация режимов кино-МРТ с исследованием внутрисердечной перфузии позволяет рассчитать градиент давления до и после места обструкции и установить степень митральной регургитации [46].
МРТ с контрастным усилением гадолинием позволяет выявить фиброз миокарда и оценить его степень. Миокардиальный фиброз можно обнаружить у 2 из 3 пациентов с ГКМП [47—49]. Гадолиний накапливается в участках фиброзированного миокарда и виден ярким участком на фоне нормального миокарда. Степень фиброза миокарда в настоящее время рассматривают как серьезный и независимый предиктор внезапной сердечной смерти [50]. В ближайшее время именно МРТ может занять лидирующие позиции в диагностике ГКМП, а также в прогнозировании течения заболевания.
Генетические аспекты диагностики

Семейная ГКМП, обусловленная появлением патологических вариантов генов саркомера сердечной мышцы, наследуется по аутосомно-доминантному типу. Гены, патологические варианты которых наиболее часто вовлечены в патогенез ГКМП, приведены в таблице Встречаемость патологии белков саркомеров миокарда при ГКМП [51, 54]. Эти гены обнаруживают у 50—60% пациентов с семейным анамнезом ГКМП, а также у 20—30% пациентов без него [52]. Примерно 6% пациентов имеют более 1 варианта саркомерного гена (являются либо гомозиготами по одному из генов, либо гетерозиготами по нескольким), и значительно меньшее число пациентов будут иметь более 1 патологического варианта всех приведенных генов [53]. Известно более 1500 определенных индивидуальных патологических вариантов генов для данной нозологии [50]. Генетическая диагностика ГКМП базируется на ряде принципов. Крайне важно собрать подробный семейный анамнез трех—четырех поколений. Особое внимание должно быть направлено на выявление у родственников любого из следующих факторов: сердечной недостаточности, ГКМП, трансплантации сердца, внезапной смерти (особенно в молодом возрасте), сердечно-сосудистых заболеваний и/или аритмий, необъяснимого инсульта, других тромбоэмболических событий. Доступны многоэлементные панели, содержащие гены, которые, как известно, связаны с ГКМП. Развитие ГКМП чаще связано с мутацией гена MYH7, кодирующего синтез миозина 7-го типа, и MYBPC3, кодирующего синтез сердечного типа миозин-связывающего белка С [51].
Возможные результаты генетического исследования: положительный, предполагающий подтверждение диагноза; отрицательный, показывающий лишь то, что из предложенных в данной панели генов ни один не вовлечен в патогенез у данного пациента; вариант неизвестного значения — лаборатория обнаружила аллельный вариант, патологичность которого вызывает сомнение, следовательно, необходимо более детальное изучение данного случая с целью верификации значимости данного варианта.
Важно отметить, что в современной клинической практике принято считать, что родственники больного c ГКМП, у которых не было обнаружено патологических вариантов какого-либо из генов, приведенных выше, не нуждаются в дальнейшем наблюдении кардиолога [44].
Консервативное лечение
Пациентам с ГКМП рекомендуется придерживаться определенного образа жизни, а именно: избегать дегидратации, приема нитратов, поддерживать оптимальную массу тела [44]. Относительно медикаментозной терапии, Американская ассоциация кардиологов рекомендует использовать неселективные адреноблокаторы (бисопролол) и недигидропиридиновые блокаторы кальциевых каналов (верапамил) [52]. Часть больных c ГКМП нуждаются в имплантации кардиовертера-дефибриллятора. Показаниями для установки данного устройства служат перенесенный инфаркт миокарда, зафиксированная фибрилляция желудочков или гемодинамически значимая желудочковая тахикардия. Стоит рекомендовать пациентам имплантацию кардиовертера-дефибриллятора при максимальной толщине ЛЖ ≥30 мм, периодически возникающих синкопальных состояниях и в том случае, если его ближайшие родственники умерли от внезапной сердечной смерти [55].
Хирургическое лечение
Стандартом хирургического лечения пациентов с обструктивной формой ГКМП, у которых аорто-желудочковый градиент давления составляет 50 мм рт.ст. и более (в покое или при физической нагрузке), вот уже почти 50 лет является септальная миоэктомия (МЭ) [35, 52, 56]. В течение последних лет послеоперационная выживаемость после септальной МЭ составляет 99, 98 и 95% в течение 1, 5 и 10 лет соответственно [57]. Такого результата удалось добиться благодаря совершенствованию хирургических и диагностических методик, а также анестезиологического пособия и методов защиты миокарда. Стоит отметить, что результат септальной МЭ тем лучше, чем больше было выполнено хирургом данных операций, поэтому наилучшие исходы подобных операций достигаются небольшим количеством хирургов в нескольких крупнейших центрах по лечению ГКМП Северной Америки и Европы [58, 59].
В середине 90-х годов была предложена методика чрескожной алкогольной септальной аблации (АСА), которая является альтернативой миоэктомии [60]. Данная процедура заключается в введении 1—4 мл 96% этанола в септальные перфоранты передней межжелудочковой артерии, что провоцирует локальный инфаркт миокарда, способствует уменьшению толщины МЖП и снижению аорто-желудочкового градиента давления [52, 60]. Несмотря на кажущуюся простоту и эффективность АСА, результаты лечения послужили поводом для обширных дебатов, начало которым было положено сразу после широкого введения аблации в практику [61]. Основными осложнениями данной процедуры являются жизнеугрожающие желудочковые тахиаритмии [52]. В недавно опубликованном Европейском исследовании [62] было отмечено, что после АСА желудочковые тахиаритмии и фибрилляция желудочков возникают в 8 раз чаще, чем после септальной М.Э. Также было выявлено, что АСА сопровождается 10-кратным увеличением частоты полной поперечной блокады, что требует установки постоянного двухкамерного кардиостимулятора [17, 63—66].
На сегодняшний день роль АСА в лечение пациентов с обструктивной ГКМП еще не определена окончательно, что создает необходимость дальнейших исследований. Учитывая количество и серьезность осложнений, возникающих после выполнения АСА, данную методику не рекомендуется выполнять рутинно. Предпочтение стоит отдавать септальной МЭ, которая на сегодняшний день является «золотым стандартом» хирургического лечения обструктивной формы ГКМП.
Перспективные направления в диагностике и лечении ГКМП
МРТ с контрастированием является перспективной методикой для определения миокардиального фиброза. Однако эффективность данной методики зависит от наличия вокруг зоны фиброза здорового миокарда, что представляет сложность в диагностике у пациентов с диффузным фибротическим процессом. Использование режима Т1-взвешенных изображений позволяет нивелировать данное ограничение [67]. Не так давно были проведены исследования, которые показали, что использование данного режима до и после контрастирования позволяет разграничить участки диффузного фибротического поражения и нормального миокарда [68].
Ранолазин показал свою эффективность в снижении коронарогенных болей, симптомов сердечной недостаточности. Его механизм действия обусловлен ингибированием медленных натриевых каналов, что уменьшает расход энергии миокардом и таким образом сдерживает развитие ГКМП. Эффективность данного препарата была подтверждена в эксперименте на белых мышах, в то время как на человеке исследования еще не проводились.
Препарат MYK-461 является новым селективным молекулярным ингибитором АТФ-азы миозина. Механизм действия препарата основан на снижении сократимости саркомеров миокарда при отсутствии влияния на общую сократимость сердца. На сегодняшний день препарат проходит клинические испытания. Существуют результаты исследований in vitro, показавшие высокую метаболическую стабильность препарата в микросомах печени изученных видов (исследование проводилось на клеточных культурах печени мышей, крыс, собак, обезьян и человека); исследования in vivo (препарат вводился перорально и парентерально мышам, крысам, собакам и обезьянам) позволили предположить удовлетворительные фармакокинетические характеристики препарата, особенно в случае перорального применения. В том случае, если данный препарат покажет свою клиническую эффективность, он станет дополнением или хорошей альтернативой привычной консервативной терапии β-адреноблокаторами и блокаторами кальциевых каналов.
Заключение
ГКМП — патология, имеющая важное значение для современной медицины ввиду частой встречаемости среди населения и сложности выявления до проявления клинических симптомов. Механизм развития данного заболевания до конца не ясен, известна лишь часть генетических маркеров, коррелирующих с возникновением ГКМП. В связи с этим, крайне важным и перспективным выглядит развитие генетической диагностики этого заболевания, поскольку она позволит проводить полноценные скрининговые исследования для его раннего выявления.
Другими важными задачами являются разработка эффективного консервативного лечения и совершенствование хирургических методов коррекции обструктивной формы заболевания с учетом индивидуальных особенностей каждого пациента (персонифицированный подход в терапии и хирургии ГКМП).
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
Сведения об авторах
Гипертрофическая кардиомиопатия — современное состояние проблемы. Вопросы эпидемиологии и номенклатуры, генетики и патофизиологии, вариантов течения и дифференциального диагноза
Проблема изучения гипертрофической кардиомиопатии (ГКМП) по-прежнему сохраняет актуальность ввиду высокой распространенности заболевания (1:500), риска внезапной сердечной смерти (ВСС) у лиц молодого трудоспособного возраста. Предметом большого интереса являются вопросы поиска дополнительных клинических, инструментальных и генетических маркеров, факторов окружающей среды, которые способны влиять на формирование клинического варианта течения ГКМП, риск ВСС и прогноз при ГКМП. Важным вопросом, требующим дальнейшего изучения, является молекулярно-генетическая характеристика заболевания. Необходима единая номенклатура различных форм и вариантов течения ГКМП для выработки тактики ведения больных и оценки результатов многоцентровых исследований.
Эпидемиологические исследования в различных удаленных друг от друга странах мира [2, 3] показали сопоставимую распространенность гипертрофии левого желудочка (ГЛЖ) неясной этиологии, которая является наиболее типичным фенотипом ГКМП. Распространенность составляет приблизительно 0,2% всего населения (т.е. 1:500), что эквивалентно по крайней мере 600 000 человек, страдающих этим заболеванием в США [4]. Приведенная статистика заболевания среди всего населения превышает относительно малый процент диагностики ГКМП в кардиологической практике. Это означает, что в большинстве случаев болезнь протекает без симптомов и изменения средней продолжительности жизни и остается недиагностированной.
«ГКМП» является предпочтительным термином для определения этой болезни [5], однако путаница в названиях, используемых для описания заболевания, возникла давно. В ранних исследованиях для обозначения ГКМП использовали более 80 самостоятельных названий, терминов и сокращений [5]. Кроме того, номенклатура, распространенная в 60—70-х годах XX века (например, термины «идиопатический гипертрофический субаортальный стеноз» — ИГСС или «гипертрофическая обструктивная кардиомиопатия» — ГОКМП), может сбить с толку. В действительности не менее чем у 1/3 пациентов не наблюдается обструкции ни в покое, ни после физиологической провокации [6]. В настоящее время термин «ГКМП», впервые использованный в 1979 г., который включает в себя понятия нарушенной и нормальной гемодинамики, является основным для определения этой болезни [5].
Итак, ГКМП — заболевание, которое характеризуется наличием ГЛЖ, развивающейся в отсутствие заболеваний, приводящих к гипертрофии миокарда [4, 5, 7—9]. При этом пациенты, положительные по генотипу, могут быть негативными по фенотипу (без признаков выраженной гипертрофии) [10, 11]. С клинической точки зрения ГКМП обычно диагностируется при толщине стенки левого желудочка (ЛЖ) ≥15 мм. Толщина миокарда 13—14 мм по данным эхокардиографии (ЭхоКГ) считается пограничной, особенно при наличии другой убедительной информации о наличии этой патологии, например семейного анамнеза ГКМП. Для оценки гипертрофии миокарда ранее основное внимание уделялось ЭхоКГ, в настоящее время чаще используется магнитно-резонансная томография сердца (МРТ) [12], обладающая большей разрешающей способностью, что позволяет получить более точную и исчерпывающую информацию. У детей увеличение толщины стенки ЛЖ определяется как толщина стенки ≥2 допустимых отклонения выше среднего (z-оценка ≥2) по возрасту, полу или размерам тела. Однако выделена когорта больных с разнообразными клиническими проявлениями заболевания, широким диапазоном толщины миокарда в отсутствие фенотипических признаков среди членов семьи при наличии у них генотипа ГКМП [13, 14]. Этих людей обычно называют «положительными по генотипу и негативными по фенотипу», оценивая их как больных с «субклинической/латентной ГКМП». Несмотря на то что сообщается о многообразии морфофункциональных типов ГЛЖ [12, 15, 16], приблизительно у 1/3 больных ГКМП наблюдаются сегментарное/ограниченное утолщение стенки и нормальная расчетная масса миокарда левого желудочка (ММЛЖ) [12]. Для подтверждения клинического диагноза ГКМП большое значение имеют такие характерные признаки, как семейный анамнез болезни, кардиальные проявления, тахиаритмии или изменения на электрокардиограмме [7, 8].
Дифференциальный диагноз при ГКМП — очень важный этап в постановке диагноза. Выявление у больного гипертрофии миокарда обусловливает необходимость тщательного поиска возможных причин ее развития. Наиболее часто дифференциальный диагноз необходимо проводить с гипертонической болезнью и «сердцем спортсмена», являющимся следствием физиологического ремоделирования, связанного с интенсивными спортивными тренировками [17, 18]. Путаница в этом случае возникает в случае, если толщина стенки составля.
Гипертрофическая кардиомиопатия. Причины, классификация ГКМП. Клиника, диагностика и лечение гипертрофической кардиомиопатии.
Согласно клинической классификации кардиомиопатии, гипертрофическая кардиомиопатия (ГКМП) представляет собой синдром необъяснимой гипертрофии левого желудочка (ЛЖ) и/или правого желудочка (ПЖ), не связанной с нагрузкой давлением на миокард. В рамках синдрома ГКМП в 40-60% случаев выявляется первичная саркомерная ГКМП, вызванная мутациями в генах, кодирующих сократительные белки миокарда. Около 5-10% приходится на вторичные фенокопии ГКМП.
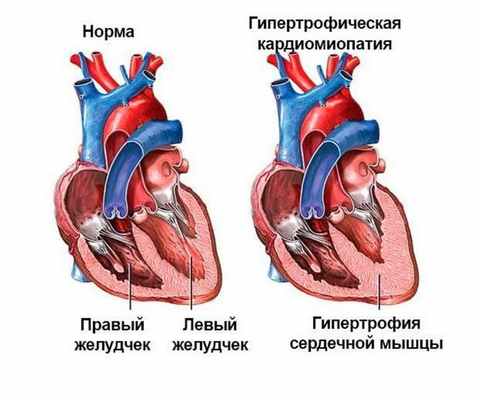
Согласно этиологической классификации, гипертрофическая кардиомиопатия - заболевание, характеризующееся необъяснимой гипертрофией недилатированного ЛЖ и/или ПЖ, чаще асимметричной, в отсутствие других системных и кардиальных заболеваний, способных привести к гипертрофии ЛЖ, наблюдаемой у конкретного больного (AHA,2011).
Распространенность ГКМП, согласно ЭхоКГ-анализу в исследовании CARDIA (1995), включавшем 4111 человек в возрасте 23-35 лет, составляет 1:500 (0,2%). Популяционный поиск патогенных ГКМП-ассоциированных саркомерных мутаций (Framingham Heart Study, Jackson Heart Study, 2012 г.) выявил их наличие у 0,6% населения, что предполагает более высокую распространенность этого заболевания, которая может составлять 1:200.
Причиной развития первичной гипертрофической кардиомиопатии является наличие мутаций как минимум в 14 известных генах сократительных белков миокарда (саркомерный комплекс), из которых наиболее часто вовлекаются тяжелые цепи β-миозина, миозинсвязывающий протеин С, тропонины Т и I, тропомиозин, актин, легкие цепи β-миозина и др. Наличие мутаций приводит к формированию хаотического, неупорядоченного положения миокардиальных волокон («disarray») с явлениями интрамиокардиального фиброза с асимметричной гипертрофией ЛЖ. Диастолическая дисфункция ЛЖ является ведущим звеном патогенеза ГКМП.
Установлено, что более половины всех случаев заболевания являются «семейными», при этом основным типом наследования является аутосомно-доминантный. Оставшуюся часть составляет так называемая спорадическая форма, когда у пациента нет родственников, страдающих ГКМП или имеющих гипертрофию миокарда. Эти случаи ГКМП вызваны случайными мутациями. Однако дифференцировка «семейной» и «спорадической» формы ГКМП бывает затруднена, так как заболевание может протекать атипично, ограничиваясь изменениями на ЭКГ.
В зависимости от наличия внутрижелудочковой обструкции - препятствия току крови в различных отделах левого желудочка (выносящего тракта левого желудочка, средней или апикальной трети ЛЖ) выделяют обструктивную и необструктивную форму ГКМП. Обструкция, в свою очередь, может быть:
■ базальной, с максимальным градиентом давления в покое ≥30 мм рт.ст.;
■ латентной, с максимальным градиентом давления в покое менее 30 мм рт.ст., но при провокационных пробах (физическая нагрузка, проба Вальсальвы, прием нитратов) градиент повышается ≥30 мм рт.ст. По характеру гипертрофии ЛЖ можно выделить:
■ классический вариант ГКМП с гипертрофией межжелудочковой перегородки, особенно базальных ее отделов;
■ апикальную ГКМП с гипертрофией верхушки ЛЖ (апикальной части межжелудочковой перегородки или анатомической верхушки ЛЖ);
■ мидвентрикулярную форму с гипертрофией средней трети ЛЖ, папиллярных мышц и внутрижелудочковой обструкцией;
■ диффузно-генерализованную форму - с гипертрофией большинства сегментов ЛЖ и уменьшением его полости.
В клинической картине ГКМП преобладают:
■ одышка при физической нагрузке, обусловленная диастолической дисфункцией ЛЖ с повышением давления в левом предсердии (ЛП) и малом круге кровообращения;
■ головокружения и обмороки при физической нагрузке, связанные с синдромом малого выброса ЛЖ из-за уменьшения его полости на фоне выраженной гипертрофии и диастолической дисфункции. Пароксизмы желудочковой тахикардии могут являться одной из причин обмороков при ГКМП;
■ нарушение ритма сердца, из которых отдельно следует рассматривать развитие фибрилляции предсердий вследствие перегрузки левого предсердия, а также неустойчивую желудочковую тахикардию, обусловленную электрической нестабильностью миокарда на фоне явлений «disarray» с наличием интрамиокардиального фиброза;
■ стенокардия напряжения, которая регистрируется в отсутствие коронарного атеросклероза и обусловлена относительной коронарной недостаточностью на фоне выраженной гипертрофии левого желудочка, диастолической дисфункцией с синдромом малого выброса, сужением коронарных артерий при гиперплазии их мышечного слоя. Наличие внутрижелудочковой обструкции усугубляет ишемию за счет увеличения нагрузки на миокард ЛЖ. Дискутабельным является вопрос о роли туннелированных коронарных артерий («мышечных мостиков»), как одной из возможных находок при ГКМП, в формировании синдрома стенокардии. Присоединение коронарного атеросклероза у больных ГКМП с синдромом стенокардии может пройти незамеченным, поэтому пациентам c ГКМП старше 40 лет с факторами риска развития ИБС рекомендуется коронароангиография для установления причины ангинозных болей;
■ кардиалгии - боли в грудной клетке, не связанные с физической нагрузкой, по длительности от 30 мин до нескольких часов;
■ внезапная сердечная смерть (ВСС) - может быть одним из проявлений ГКМП, в том числе единственным. Частота ВСС среди больных гипертрофической кардиомиопатией составляет 1-2% ежегодно. К факторам риска ВСС относятся молодой возраст пациентов (до 30 лет), семейный анамнез ВСС родственников пациента, обмороки за последние 6 мес, неустойчивая желудочковая тахикардия (не более 30 с) при суточном мониторировании ЭКГ, высокая степень гипертрофии ЛЖ (более 30 мм). Большие размеры левого предсердия и выраженная внутрижелудочковая обструкция увеличивают риск ВСС. ESC предложен калькулятор, где на основании вышеперечисленных параметров можно рассчитать ВСС за 5 лет в процентах (низкий риск до 4%, от 4 до 6% - средний, 6% и более - высокий). Наличие высокого риска ВСС является основанием для установки имплантируемого кардиовертера-дефибриллятора (ИКД), средний риск предполагает рассмотрение ИКД с учетом потенциальных факторов риска ВСС («злокачественные» генетические мутации, неадекватная реакция АД на физическую нагрузку, интрамиокардиальный фиброз на МРТ сердца с гадолинием, апикальная аневризма левого желудочка).
Саркомерный вариант ГКМП следует дифференцировать с рядом заболеваний, которые сопровождаются необъяснимой гипертрофией миокарда: болезнями накопления (гликогенозы: PRKAG 2 и Данон-тип, болезнь Андерсона-Фабри), инфильтративными заболеваниями (амилоидоз, болезнь Гоше, синдром Гутлера и др.), гипертоническим сердцем, «спортивным сердцем», митохондриопатиями, нейромышечными заболеваниями и синдромами мальформаций в педиатрической практике. Нередко лабораторные тесты позволяют заподозрить специфическую природу фенокопий ГКМП:
■ снижение СКФ и протеинурия может наблюдаться при амилоидозе, болезни Андерсона-Фабри и болезнях митохондриальной дезоксирибонуклеиновой кислоты (ДНК);
■ печеночные показатели могут быть изменены при митохондриальной патологии, болезни Данона и дефектах β-окисления;
■ сывороточная креатининфосфокиназа будет повышена при метаболической патологии, такой как болезнь Данона или митохондриальная болезнь;
■ низкий уровень α-галактозидазы А в плазме/лейкоцитах выявляется при болезни Андерсона-Фабри;
■ определение легких цепей иммуноглобулинов в сыворотке, иммунофиксация плазмы и мочи, электрофорез мочи позволяют выявить амилоидоз;
■ глюкоза натощак может быть повышена при некоторых болезнях митохондриальной ДНК и понижена при болезнях обмена жирных кислот и карнитина;
■ лактат плазмы увеличен у некоторых больных с митохондриальными заболеваниями.
Показано ограничение физической нагрузки, однако обязательно должны выполняться адекватные состоянию физические упражнения. Запрещается профессиональное занятие спортом и участие в соревнованиях.
Основу терапии ГКМП составляют препараты с отрицательным хронотропным действием.
Невазодилатирующие β-АБ (пропранолол, атенолол, бисопролол, метопролол) с подбором максимально переносимой дозы рекомендуются в качестве препаратов первой линии для уменьшения симптомов.
Верапамил с подбором максимально переносимой дозы рекомендуется при непереносимости или противопоказаниях к приему β-АБ.
Дилтиазем с подбором максимально переносимой дозы рекомендуется при непереносимости или противопоказаниях к приему β-АБ и верапамила для облегчения симптомов.
Низкие дозы петлевых или тиазидных диуретиков могут использоваться с осторожностью для уменьшения одышки при недостаточном эффекте β-АБ или верапамила.
При отеке легких и гипотензии на фоне внутрижелудочковой обструкции рекомендуются пероральные или внутривенные β-АБ.
При наличии ХСН со сниженной ФВ ЛЖ ( <50%) ИАПФ/блокаторы рецепторов ангиотензина (БРА) должны рассматриваться в дополнение к β-АБ.
Антагонисты рецепторов к минералокортикоидам должны рассматриваться при II-IV ФК ХСН по классификации Нью-Йоркской ассоциации сердца (NYHA), несмотря на лечение ИАПФ/ БРА (рис. 1.1).
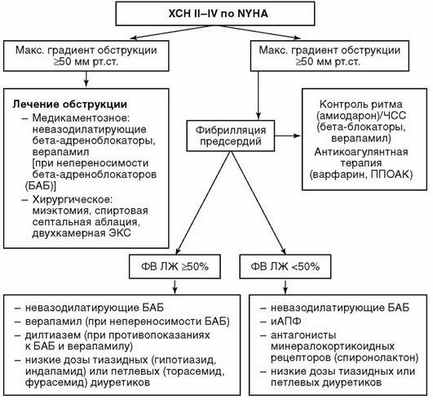
Рис. 1.1. Алгоритм лечения хронической сердечной недостаточности при гипертрофической кардиомиопатии
При наличии пароксизмальной/постоянной формы фибрилляции предсердий антикоагулянты [варфарин под контролем международного нормализованного отношения (МНО) 2-3, прямые пероральные антикоагулянты: дабигатрана этексилат, ривароксабан, апиксабан] назначаются всем пациентам с ГКМП без учета шкалы CHA2DS2VASc.
Восстановление синусового ритма при фибрилляции предсердий проводится с помощью электроимпульсной терапии (ЭИТ) или внутривенного введения амиодарона. Амиодарон рассматривается как основной препарат для поддержания синусового ритма. β-АБ, верапамил или дилтиазем рекомендуются для контроля ЧСС при наличии постоянной формы фибрилляции предсердий. Аблация АВ-узла с имплантацией электрокардиостимулятора (ЭКС) выполняется при неэффективном контроле ЧСС с помощью медикаментозной терапии и прогрессировании ХСН на фоне фибрилляции предсердий.
Для пациентов с плохо переносимой желудочковой тахикардией должны рассматриваться установка ИКД и лечение β-АБ и амиодароном.
Важно помнить, что препараты с вазодилатирующим эффектом, нитраты и симпатомиметики противопоказаны при ГКМП, особенно при наличии внутрижелудочковой обструкции.
Хирургические методы лечения гипертрофической кардиомиопатии включают:
■ миэктомию;
■ транскатетерную спиртовую септальную аблацию межжелудочковой перегородки;
■ двухкамерную стимуляцию с укороченной АВ-задержкой;
■ ИКД при высоком и среднем риске ВСС.
Читайте также:
- Топография мочевого пузыря. Мочевой пузырь. Строение мочевого пузыря.
- Лучевые признаки доброкачественной метастазирующей миомы
- Пленчато-язвенная ангина Пло-Винцента (Plaut-Vincent). Флегмонозная ангина (перитонзиллярный гнойник)
- Сущность гаметогенеза. Цитология гаметогенеза - мейоз
- ЭхоКГ при рестриктивной кардиомиопатии