Конечностно-поясная дистрофия
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Конечностно-поясная мышечная дистрофия. Диагностика
Конечностно-поясная мышечная дистрофия - это термин охватывает группу прогрессирующих наследственных миопатий, при которых поражаются главным образом мышцы тазового и плечевого пояса В конечном итоге также развивается атрофия и слабость мышц дистальных отделов конечностей. При некоторых формах формируется гипертрофия икроножных мышц и контрактура голеностопных суставов, в связи с чем может быть ошибочно диагностирована мышечная дистрофия Беккера.
Первые клинические проявления коненостно-поясной мышечной дистрофии редко возникают до среднего или старшего детского возраста либо могут быть отсрочены до раннего зрелого возраста. Боль в нижней части спины может служить первой жалобой и обусловлена формированием лордоза в связи со слабостью ягодичных мышц. Потеря способности к ходьбе (больные прикованы к инвалидному креслу) наблюдается обычно около 30 лет. Скорость прогрессирования заболевания варьирует в разных поколениях одной семьи, но одинакова у пациентов одного поколения.
Характерна слабость мышц-сгибателей и -разгибателей шеи, однако поражение мышц лица, языка и других мышц бульбарной группы встречается редко. По мере прогрессирования мышечной слабости и атрофии сухожильные рефлексы снижаются. Поражение сердца нехарактерно. Интеллект, как правило, не снижен. Дифференциальный диагноз конечностно-поясной мышечной дистрофии следует проводить с ювенильной спинальной мышечной атрофией (болезнь Кугельберга—Веландер), миастенией и метаболическими миопатиями:
В большинстве случаев конечностно-поясная мышечная дистрофия наследуется по аутосомно-рецесивному типу, в некоторых семьях наблюдается аутосомно-доминантный тип наследования. В последнем случае заболевание часто характеризуется доброкачественным течением и функциональные нарушения выражены незначительно.

ЭМГ и мышечная биопсия подтверждают диагноз мышечной дистрофии, но ни один из этих методов не специфичен настолько, чтобы установить точный диагноз без дополнительных клинических критериев. В некоторых случаях выявляется дефицит адхалена — дистрофинассоциированного гликопротеида сарколеммы; этот специфический дефект можно определить иммуноцитохимически на материале мышечной биопсии. Характерно повышение активности КФК в крови, однако она варьирует в разных семьях. ЭКГ, как правило, без изменений.
При одной аутосомно-доминантной форме конечностно-поясной мышечной дистрофии генетический дефект локализован на длинном плече хромосомы 5. При аутосомно-рецессивной форме ген картирован на длинном плече хромосомы 15. Продукт мутантного гена — дистрофинассоциированный протеин в саркогликановом комплексе (саркогликанопатия) ответствен за развитие некоторых случаев конечностно-поясной мышечной дистрофии с аутосомно-рецессивным типом наследования. Адхален — это а-саркогликан; встречаются другие формы конечностно-поясной мышечной дистрофии, обусловленные дефицитом b-, у- и q-саркогликана.
В нормальных (неизмененных) гладких мышцах а-саркогликан замещается на е-саркогликан.
Другая группа конечностно-поясных мышечных дистрофий обусловлена аллельными мутациями гена дисферлина (DYSF). Аномальный ген отвечает за синтез протеина, необходимого для структурной целости сарколеммы, хотя и не ассоциированного с комплексом дистрофин-гликопротеид. DYSF взаимодействует с кавеолином-3 или калпаином-3; дефицит DYSF может быть вторичным по отношению к дефектам этих продуктов других генов. Описаны формы с аутосомно-рецессивным (миопатия Миоши) и аутосомно-доминантным типом наследования.
Обе формы миопатий характеризуются медленно прогрессирующим течением с дебютом в подростковом или молодом возрасте и возможным поражением как дистальных, так и проксимальных мышц. Кардиомиопатия развивается редко. При дисферлинопатиях обнаружено повышение активности КФК в крови в несколько тысяч раз. Ультраструктурные изменения включают утолщение базальной пластинки над дефектами сарколеммы и замещение сарколеммы множественными слоями, состоящими из мелких пузырьков. Количество регенерирующих мышечных волокон превосходит таковое мышечных волокон с признаками дегенеративных изменений.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Мышечная дистрофия поясно-конечностная, FKRP м.
FKRP (FUKUTIN-RELATED PROTEIN) - фукутин-связанный белок. Расположен на хромосоме 19 в области 19q13.32. Содержит 3 некодирующих экзона и один большой кодирующий экзон.
Мутации в данном гене приводят также к развитию таких заболеваний как поясно-конечностная мышечная дистрофия тип 5А (с аномалиями мозга и глаз) и поясно-конечностная мышечная дистрофия тип 5В (с или без умственной отсталости.
Относится к группе прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового поясов конечностей.
Клинические проявления заболевания отличаются значительным разнообразием с наличием как внутрисемейного, так и межсемейного полиморфизма. Описаны как пациенты с ранним началом, тяжелым протеканием и быстрой инвалидизацией, так и началом до 40 лет и медленным прогрессированием. Типичными клиническими проявлениями этой группы заболевания являются нарушения походки (переваливающаяся или «утиная» походка), «осиная» талия, приемы Говерса (подъем лесенкой из положения на корточках), гиперлордоз в поясничном отделе позвоночника, «крыловидные» лопатки, симптом «дряблых надплечий» (при попытке приподнять больного за подмышечные области, его плечи свободно поднимаются вверх, а голова как бы проваливается между ними), сухожильная гипорефлексия, мышечная гипотония и гипотрофия. Мышцы лица, как правило, не поражаются. Слабость проксимальных мышц может начаться либо в верхних конечностях, либо в нижних, но обычно при прогрессировании патологического процесса поражаются все четыре конечности. Мышечная слабость может проявиться уже в детстве (в возрасте до пяти лет) или же позже: например, на 3-м десятилетии жизни; иногда она сопровождается псевдогипертрофией икроножных и других мышц, контрактуры выявляются редко. После первых проявлений заболевания больной может сохранить способность ходить более 20 лет. Мужчины и женщины болеют в равной мере. Некоторые врачи отмечают, что если болезнь появилась в детстве, то она прогрессирует быстрее и разрушительнее; если начало пришлось на юность или взрослый возраст, то обычно болезнь прогрессирует медленнее. У некоторых больных в патологический процесс вовлекается сердце, но это бывает не так часто, как при других видах миодистрофий. Проблемы с сердцем могут проявиться в виде кардиомиопатий (слабость сердечной мышцы) или аритмий. В отдельных случаях заболевание само по себе может проявляться как кардиомиопатия. По истечении тридцати или более лет к основным признакам болезни присоединяется дыхательная недостаточность. Боль не является характерной составляющей болезни. Но ограниченная подвижность иногда приводит к болезненности мышц и суставов. Интеллектуальные функции не страдают. Для большинства нозологических форм поясно-конечностных прогрессирующих мышечных дистрофий характерно повышение активности креатинфосфокиназы в плазме крови больных, которое может выявляться еще на доклинической стадии. Увеличение активности этого фермента может выявляться и у гетерозиготных носителей мутации в том или ином гене, которое, однако, не достигает такой степени, как у больных. На ЭМГ и в мышечном биоптате выявляются признаки миопатии, мышцы истончены, часть волокон замещена жировой и соединительной тканью. В саркоплазме выявляются очаги фокального некроза. Ядра мышечных волокон центрально смещены, располагаются рядами или цепочками, вакуолизированы, с выраженным ядрышком. На поздних стадиях волокна теряют поперечную исчерченность, фрагментированы; иногда обнаруживаются только остатки миофибрилл. При дифференциальной диагностике необходимо исключить воспалительные и метаболические миопатии, а также фенотипически сходные спинальные мышечные атрофии.
Частота встречаемости: Частота всех поясно-конечностных мышечных дистрофий колеблется в различных популяциях от 5 до 70 больных на 1 миллион населения.
Мышечная дистрофия поясноконечностная
В 1954 году доктором Уалтоном и доктором Натрассом был впервые введен термин поясно-конечностые мышечные дистрофии (ПКМД илиLGMD), однако четкого понимания механизма заболевания не было. Это был универсальный термин, который использовали достаточно широко для разграничения людей с преобладающей конечностно-поясной слабостью от людей с другими типами дистрофий, таких, как Дюшена, Ландузи-Дежерина и др.
Поясно-конечностные мышечные дистрофии (ПКМД) - группа прогрессирующих мышечных дистрофий, для которых характерно изолированное или преимущественное поражение мышц плечевого и тазового поясов конечностей. Типичными клиническими проявлениями этой группы заболевания являются нарушения походки (переваливающаяся или «утиная» походка), «осиная» талия, приемы Говерса (подъем лесенкой из положения на корточках), гиперлордоз в поясничном отделе позвоночника, «крыловидные» лопатки, симптом «дряблых надплечий» (при попытке приподнять больного за подмышечные области, его плечи свободно поднимаются вверх, а голова как бы проваливается между ними), сухожильная гипорефлексия, мышечная гипотония и гипотрофия. Мышцы лица, как правило, не поражаются. Слабость проксимальных мышц может начаться либо в верхних конечностях, либо в нижних, но обычно при прогрессировании патологического процесса поражаются все четыре конечности. Мышечная слабость может проявиться уже в детстве (в возрасте до пяти лет) или же позже: например, на 3-м десятилетии жизни; иногда она сопровождается псевдогипертрофией икроножных и других мышц, контрактуры выявляются редко.
После первых проявлений заболевания больной может сохранить способность ходить более 20 лет. Мужчины и женщины болеют в равной мере. Некоторые врачи отмечают, что если болезнь появилась в детстве, то она прогрессирует быстрее и разрушительнее; если начало пришлось на юность или взрослый возраст, то обычно болезнь прогрессирует медленнее.
У некоторых больных в патологический процесс вовлекается сердце, но это бывает не так часто, как при других видах миодистрофий. Проблемы с сердцем могут проявиться в виде кардиомиопатий (слабость сердечной мышцы) или аритмий. В отдельных случаях заболевание само по себе может проявляться как кардиомиопатия.
По истечении тридцати или более лет к основным признакам болезни присоединяется дыхательная недостаточность. Боль не является характерной составляющей болезни. Но ограниченная подвижность иногда приводит к болезненности мышц и суставов. Интеллектуальные функции не страдают. Для большинства нозологических форм поясно-конечностных прогрессирующих мышечных дистрофий характерно повышение активности креатинфосфокиназы в плазме крови больных, которое может выявляться еще на доклинической стадии. Увеличение активности этого фермента может выявляться и у гетерозиготных носителей мутации в том или ином гене, которое, однако, не достигает такой степени, как у больных.
На ЭМГ и в мышечном биоптате выявляются признаки миопатии, мышцы истончены, часть волокон замещена жировой и соединительной тканью. В саркоплазме выявляются очаги фокального некроза. Ядра мышечных волокон центрально смещены, располагаются рядами или цепочками, вакуолизированы, с выраженным ядрышком. На поздних стадиях волокна теряют поперечную исчерченность, фрагментированы; иногда обнаруживаются только остатки миофибрилл.
При дифференциальной диагностике необходимо исключить воспалительные и метаболические миопатии, а также фенотипически сходные спинальные мышечные атрофии.
Частота всех поясно-конечностных мышечных дистрофий колеблется в различных популяциях от 5 до 70 больных на 1 миллион населения.В зависимости от типа наследования поясно-конечностные мышечные дистрофии разделяют на два типа.
К 1 типу относят нозологические формы с аутосомно-доминантным типом наследования. Для всех заболеваний этой группы известна локализация генов на хромосомах и для трех из них идентифицированы белковые продукты.
Ко второму типу относят формы заболевания, наследующиеся аутосомно- рецессивно. Первое описание ПКМД с аутосомно-рецессивным типом наследования проведено Kloepfer и Tally в 1958 году. Для большинства заболеваний этой группы идентифицированы гены, описаны основные типы их патологических мутаций и известен белковый продукт и его основные функции. На данный момент выделено 16 нозологических форм ПКМД с аутосомно-рецессивным наследованием и 7 с аутосомно-доминантным (Таблица 1), и их поиск продолжается.
Аутосомно-рецессивные формы ПКМД являются более распространенными, чем аутосомно- доминантные, которые составляют около 10% всех ПКМД. Различные группы населения часто имеют разные частоты различных типов ПКМД. Среди аутосомно-рецессивных форм наиболее распространены ПКМД 2A (от 30 % в Бразилии до 80% среди басков Испании от всех выявленных случаев) и ПКМД 2B (от 20% до 40% всех случаев). На группу саркогликанопатий (ПКМД 2C-2F) приходится еще около 20-25% всех случаев ПКМД, эта группа характеризуется тяжелым течением заболевания. Как и в случае с другими ПКМД, различные саркогликанопатии с различной частотой встречаются в разных популяциях. ПКМД 2C широко распространена в Тунисе; ПКМД 2D распространена в Европе, Соединенных Штатах и Бразилии, а ПКМД 2E и ПКМД 2F широко распространены в Бразилии. ПКМД 2I довольно распространена, особенно среди жителей Северной Европы. Недавние исследования показали, что больные с мутациями в этом гене составляют 6-38% случаев ПКМД. Остальные аутосомно-рецессивные формы ПКМД встречаются редко, и часто наблюдаются в изолированных группах населения.
В ООО «Центр Молекулярной Генетики» методом прямого автоматического секвенирования всей кодирующей последовательности проводится диагностика следующих форм ПКМД (LGMD1A, LGMD1B, LGMD1С, LGMD2А, LGMD2С, LGMD2D, LGMD2E, LGMD2F, LGMD2К, LGMD2I, LGMD2М, LGMD2L). Данные формы выделены в таблице синим цветом.
Для LGMD 2I типа описаны частые мутации с.826C>A, с.229С>T, которые встречаются хотя бы на одной из хромосом более чем у 80% больных из РФ.
Для LGMD 2A типа описаны частые мутации с.550delA, с.598_612del15, которые встречаются хотя бы на одной из хромосом более чем у 83% больных из РФ.
По литературным данным для LGMD 2D типа описаны частые мутации, характерные для больных из РФ: c.229C>T и c.271G>A.
По литературным данным для LGMD 2L типа описаны частые мутации, характерные для европейцев: c.191dupA и c.2272C>T.
В ООО «Центр Молекулярной Генетики» проводится анализ частых мутаций с.826С>A, с.229С>T и замены с.341С>T гена FKRP, c.191dupA и c.2272C>T гена ANO5 и c.229C>T и c.271G>A гена SGCA , с.550delA и с.598_612del15 гена CAPN3.
Конечностно-поясная дистрофия
Вниманию обучающихся! Приём документов для участия в конкурсе на соискание повышенной государственной академической стипендии проводится.
Внимание поступающих в ординатуру, обучавшихся по программам специалитета в образовательных организациях с заключением договора о целевом.
I Всероссийский конкурс студенческих проектов «Октябрь - месяц охраны психического здоровья» Союз охраны психического здоровья объявляет о.
Приглашаем Вас принять участие во II межрегиональной научно-практической конференции «Современные лечебно-диагностические технологии в хирургии.
Эл. почтовый ящик:
Эксклюзивная лекция «Конечно-поясная мышечная дистрофия. Алгоритмы для врачей»
Эксклюзивная лекция «Limb Girdle Muscular Dystrophies. An algorithm for clinicians» – «Конечно-поясная мышечная дистрофия. Алгоритмы для врачей» доктора Джона Уртизбереа Андони состоится 14 августа 2017 года на базе кафедры неврологии ФПК и ППС Дагестанского государственного медицинского университета. Начало лекции в 9:00.
Доктор Джон Уртизбереа Андони – педиатр, главный консультант отделения нервно-мышечной патологии в госпитале Энде (Hendaye), заместитель директора Центра обращения больных с нервно-мышечной патологией Энде (Франция), руководитель учебного курса «Школа миологии» в Институте Миологии (г. Париж, Франция).
Andoni Urtizberea является руководителем всемирно известной Парижской летней школы Миологии и консультантом в нервно-мышечном справочном центре в больнице Энде во Французском Баскском регионе. В Энде он отвечает за клинику, предназначенную исключительно для нервно-мышечных пациентов (150-200 и взрослых, 10% из которых приезжают из соседней Испании). Отделение активно связывается с другими французскими подразделениями, посвященными нервно-мышечным заболеваниям.
Ежегодно Парижская летняя школа Миологии, которую он основал в 1997 году вместе с Мишелем Фардо, обучила 400 студентов из 55 национальностей за 10 лет и признана ведущей специализированной школой миологии. Более поздняя разработка – Escuela de Verano Latino-Americana de Miologia (EVELAM), летняя школа на испанском языке, которая проводится каждый год в Латинской Америке, была начата в Чили (Сантьяго, 2008 г.) и с тех пор прошла в Уругвае (Монтевидео, 2009 г.) и Аргентике (Кордова, 2010 г.). Каждый курс в среднем объединяет 100-120 участников.
Андони также выступает в качестве клинического директора Русской весенней школы миологии (Санкт-Петербург, 2010 г., Москва, 2011 г.) и участвует в организации специальных мероприятий, в том числе трехгодичных конгрессов Миологии во Франции. В результате своей веры в важность продолжения и поддержания связей, разработанных во время учебных инициатив, Андони разработал инновационную систему телемедицины в области миологии и ежегодно получает около 200 докладов о случаях заболевания выпускников Летней школы в Париже. Это позволяет поддерживать надежные связи со многими командами по всему миру.
Если вы нашли ошибку, пожалуйста, выделите фрагмент текста и нажмите Ctrl+Enter.
Мышечная дистрофия
Мышечная дистрофия (миодистрофия) — это целая группа наследственных заболеваний, для которых характерны слабость и дегенерация мышц скелета. Подобные патологии обычно протекают без болевых ощущений и потери чувствительности. Пораженные недугом мышцы увеличиваются в размерах, ложно создавая впечатление окрепшей мускулатуры. Наиболее распространенные миодистрофии — Эрба-Рота, Эмери-Дрейфуса, Ландузи-Дежерина и дистрофия Беккера.
Выделяют несколько разновидностей мышечной дистрофии:
- Прогрессирующая Дюшена — составляет 50% от общего числа заболеваний. Патология характерна для лиц мужского пола. Все дело в Х-половой хромосоме, которая передается от матери к сыну. Девочки, в свою очередь, являются носителями.
- Прогрессирующая дистрофия Беккера — для такой формы характерно более медленное развитие с не ярко выраженной симптоматикой.
- Плече-лопаточно-лицевая миопатия (Ландузи-Дежерина) — поражает мужчин и женщин. Наследуется по аутосомно-доминантному, аутосомно-рецессивному или Х-связанному типу.
- Конечностно-поясная — порядка 30% мутаций возникают вследствие фенотипных мутаций генов.
Причины возникновения
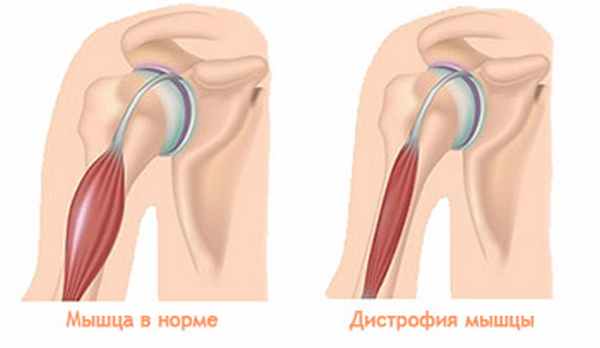
Как было сказано выше, основная причина возникновения мышечной дистрофии — патологии в половых генах. Медицине известно порядка 25 мутаций, которые становятся причиной нарушения процесса выработки белка дистрофина. Он отвечает за нормальную работу мышечных волокон. Недостаток дистрофина приводит к повреждению клеток и истощению мембраны.
Симптомы и признаки
Для мышечной дистрофии характерны следующие симптомы:
Симптомы прогрессирующей мышечной дистрофии:
- снижение тонуса в мышцах;
- атрофия скелетных мышц;
- нарушения в походке и частые падения;
- трудности при беге или быстрой ходьбе;
- снижение или полная утрата физических навыков;
- увеличение мышц в размерах;
- усталость и общий упадок сил;
- разрастание мышечной ткани на месте разрушенного волокна.
Какой врач лечит
Лечением мышечной дистрофии занимается врач-невролог. Специалист этого профиля подберет наиболее точный способ диагностики и назначит подходящую терапию. В нашей клинике работают опытные врачи, которые распознают миодистрофию на ранней стадии и примут все необходимые меры.
Методы диагностики
Определить мышечную дистрофию возможно несколькими способами. Чаще всего врачи назначают комплекс диагностических мероприятий:
- анализ присутствия фактов мышечной дистрофии в семье;
- электромиография;
- исследование участка пораженной мышцы под микроскопом;
- электромиография;
- исследования иммунитета;
- МРТ мягких тканей.
Магнитно-резонансная терапия — наиболее информативный метод метод. Под действием радиоволн и магнитного поля получаются снимки, позволяющие идентифицировать пораженную мышцу и степень ее разрушения.
Методы лечения
Мышечная дистрофия успешно лечится, если вовремя принять меры. Невролог выбирает схему в зависимости от формы заболевания и степени тяжести. Полностью избавиться от заболевания не получится, но облегчить симптоматику и предупредить осложнения реально.
Комплексный подход к лечению мышечной дистрофии включает в себя следующие этапы:
- Назначение кортикостероидов;
- Прием анаболических гормонов;
- Включение в ежедневный рацион БАДов;
- Занятие ЛФК;
- Массаж;
- Сбалансированная диета.
Результаты
Правильно назначенное лечение и своевременная диагностика дистрофии гарантируют избавление от симптоматики и предупреждение развития осложнений. Именно по этой причине важно обратиться к врачу при первых клинических проявлениях заболеваниях, а также в том случае, если в роду имели место быть подобные случаи.
Реабилитация и восстановление образа жизни
Самый негативный прогноз у пациентов с дистрофией Дюшена. В остальных случаях дальнейшее развитие зависит от формы патологии и других факторов. Больному следует придерживаться врачебных рекомендаций, избегать изнурительных физических нагрузок, вести здоровый образ жизни и соблюдать назначенные врачом дозировки.
Образ жизни при мышечной дистрофии
Своевременное обращение к врачу для проведения обследования, умеренная физическая активность и правильное питание помогут пациенту продлить состояние ремиссии. При успешном лечении больной может полностью избавиться от симптоматики и вернуться к привычному образу жизни, но с определенными ограничениями. Пациентам с диагнозом мышечная дистрофия потребуется скорректировать рацион, обогатить его витаминами и избавиться от вредных привычек.
Читайте также:
- Травмы почек
- Патогенез лабиринтопатии при шейном остеохондрозе. Ангиовертебронгенная патология лабиринта
- Добросовестность ученых. Исследовательская недобросовестность
- Эндотелий сосудов при нестабильной стенокардии. Миокард при нестабильной стенокардии
- Стробоскопия гортани. Методика ларингостробоскопии