Краниосиностоз
Добавил пользователь Alex Обновлено: 27.01.2026
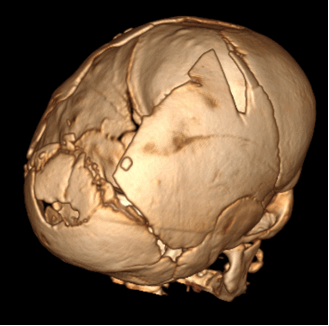
Заболевание характеризуется КС коронарных швов с формированием брахицефалии или акроцефалии, вдавленной деформацией средней зоны лица и симметричной синдактилией конечностей. Частота возникновения СА составляет от 7,6 до 22 случаев на 1 млн живорожденных, причем у азиатов частота встречаемости самая высокая, а у испанцев – самая низкая. Наследование СА происходит по аутосомно- доминантному типу и вызывается мутацией гена рецептора фактора роста фибробластов 2-го типа (fibroblast growth factor receptor 2 – FGFR2), расположенного на длинном плече хромосомы 10. Одной из отличительных особенностей пациентов с СА является макрокрания, которая сочетается с КС.
Другой особенностью СА является преждевременное смыкание сфеноокципитального и петроокципитальных синхондрозов.
Нередко у пациентов обнаруживают пороки развития головного мозга, такие как дистопия миндалин мозжечка, стеноз яремного отверстия, арахноидальные кисты в задней черепной ямке, мальформации мозолистого тела и/или лимбических структур.
Следует заметить, что прогрессирующая гидроцефалия неспецифична для заболевания, за нее ошибочно принимают непрогрессирующую вентрикуломегалию, часто наблюдаемую у этих больных. У пациентов с СА обычно наблюдается умственное недоразвитие разной степени выраженности, однако имеются пациенты и с нормальным интеллектом. К основным лицевым признакам СА относится окулярный проптоз (экзорбитизм), который может быть асимметричным. Поражения органа зрения. Гипоплазия верхней челюсти,расщелина язычка или мягкого нёба, расщелина твердого нёба, альвеолярного отростка верхней челюсти и губы очень редки. Задержка прорезывания зубов. Кроме описанных пороков развития, примерно у 2/3 пациентов наблюдается срастание шейных позвонков. Также описаны пороки развития трахеи.
Синдром Крузона (Crouzon syndrome)
Синдром Крузона (СК) является типичным КС с вовлечением не только коронарного, но и сагиттального и лямбдовидного швов. Порок сопровождается выраженной гипоплазией средней трети лица, с очевидным окулярным проптозом, но при этом заболевании не выявляют грубых пороков развития кистей и стоп, что отличает его отдругих заболеваний этой группы. СК является самым частым в группе синдромальных КС и наблюдается у 1 из 65 000 новорожденных. Так же как и СА он наследуется по аутосомно-доминантному типу и вызывается мутацией гена FGFR2. Обычно к моменту рождения уже синостозировано несколько швов черепа, а в течение первых лет жизни количество включенных в процесс швов может увеличиться. Преждевременное смыкание сфеноокципитального и петрозоокципитального синхондрозов при СК наблюдается часто и происходит в конце внутриутробного периода или вскоре после рождения. Форма головы зависит от того, какие швы и в какой последовательности синостозировались. Кости черепа обычно тонкие, с отчетливыми пальцевыми вдавлениями. Передняя, средняя и задняя черепные ямки короткие, но при этом в отличие от СА практически всегда симметричные. Пороки развития ЦНС наблюдаются у пациентов с СК реже, чем при СА. Они включают аномалию Киари 1-го типа и прогрессирующую гидроцефалию. Стеноз яремных отверстий в сочетании с сужением яремных вен наблюдается в 60% случаев. Более выраженный симметричный экзорбитизм. Верхняя челюсть сильно недоразвита, что в сочетании с костным дефицитом нижнего края орбит усиливает окулярный проптоз. Нёбо узкое, высокое, у половины больных отмечаются латеральные утолщения слизистой оболочки. Нарушения окклюзии и скученность зубов на фоне гипоплазии верхней челюсти так же являются характерными признаками порока, однако в отличие от СА значительной задержки прорезывания зубов нет. Сращение шейных позвонков у 22% больных. Примерно у половины — кондуктивная тугоухость. У 13% имеется стеноз или атрезия наружного слухового канала. Часто наблюдаются значительные нарушения дыхания.
Синдром Пфайффера (Pfeiffer syndrome)
Типичными проявлениями синдрома Пфайффера (СП) являются коронарный КС, гипоплазия верхней челюсти, экзорбитизм и пороки развития конечностей, такие как широкие большие пальцы рук и ног, синдактилия и брахидактилия. При СП выявляются мутации генов FGFR1 или FGFR2. Наследуется синдром по аутосомнодоминантному типу. Частота синдрома не определена из-за его редкости. Выделяют три типа синдрома.
Тип I описывается как классическая или мягкая форма заболевания. Он характеризуется КС коронарного и нередко сагиттального шва, с формированием брахицефалии. Деформация черепа и лица при этом типе напоминает таковую при мягком фенотипе СА. Пациенты с этим типом обычно имеют нормальный или близкий к норме уровень интеллекта и обычную продолжительность жизни.
Тип II более тяжелый и характеризуется КС многих швов. В основном зарастают коронарный, лямбдовидный и метопический швы с компенсаторным ростом черепа по линии открытых сагиттального и височно-теменных швов, что придает голове форму трилистника (череп в виде листа клевера – Cloverleaf skull). При этом типе может отмечаться тяжелый экзорбитизм. Интеллект у детей часто снижен, одним из характерных признаков является плечелучевой синостоз. При отсутствии лечения продолжительность жизни детей с этим типом СП невелика.
Тип III по тяжести черепно-лицевых проявлений похож на тип II, за исключением того, что КС не приводит к формированию трехдольчатого черепа. Большинство пациентов фенотипически напоминают СК, от которого их отличают характерные деформации больших пальцев руки ног. Также у них наблюдается плечелучевой синостоз. Характерными для всех типов СП являются типичные деформации больших пальцев рук и ног в виде их укорочения, утолщения и клинодактилии. Другими признаками являются кожная синдактилия, чаще пальцев стоп и брахидактилия кистей и стоп за счет укорочения средних фаланг. Все пациенты с СП имеют гипоплазию средней зоны лица в сочетании со скученностью зубов и нарушениями прикуса.
Синдром Джексона–Вейса (Jackson–Weiss syndrome)
Синдром Джексона–Вейса характеризуется вовлечением в патологический процесс нескольких швов черепа с формированием акроцефалии. Гипоплазия средней трети лица также типична для этого заболевания, но выражена не так сильно, как при СК. Еще одним характерным признаком являются утолщение и девиация больших пальцев стоп, как при СП, большие пальцы рук при этом не поражаются. Наследуется заболевание по аутосомно-доминантному типу с вариабельной экспрессивностью. Мутация происходит в гене FGFR2. Интеллектуальное развитие часто не страдает. Хотя и описаны индивидуумы с низким или пограничным уровнем интеллекта.
Синдром Карпентера (Carpenter syndrome)
Типичными признаками синдрома Карпентера являются КС и полисиндактилия стоп. В отличие от СА, при котором также описана постаксиальная полидактилия (полидактилия мизинца), при синдроме Карпентера полидактилия всегда преаксиальная. Череп при этом заболевании деформирован по типу акроцефалии и не имеет характерных отличий от других синдромальных КС. Заболевание наследуется по аутосомно-рецессивному типу и его связывают с дефектом RAB23 из группы Ras-генов, который является негативным регулятором сигнальной системы группы НН (hedgehog). Другими признаками болезни являются низкий рост, ожирение, врожденные пороки сердца, задержка интеллектуального развития. Выбор тактики и сроков лечения напрямую зависит от тяжести внутричерепной гипертензии.
Синдром Сэтре–Чотзена (Saethre–Chotzen syndrome)
Синдром Сэтре–Чотзена (ССЧ) единственный из описанных выше синостозов характеризуется асимметрией черепа и лица. Основные проявления ССЧ включают КС, птоз верхних век, высокий лоб с низкой линией роста волос и пороки развития конечностей, такие как брахидактилия и мягкотканная синдактилия. ССЧ наследуется по аутосомно-доминантному типу с высокой пенетрантностью и обусловлен главным образом мутацией гена TWIST. КС наблюдаются у большинства, но не у всех пациентов, чаще всего срастается коронарный шов, приводя к брахицефалии или акроцефалии. Также отмечено зарастание лямбдовидного и метопического швов. Для ССЧ характерны позднее закрытие родничков, увеличенные отверстия теменных выпускников и другие дефекты костей черепа. Длина передней черепной ямки укорочена, а турецкое седло может быть углублено. Из лицевых признаков ССЧ отмечают широкое уплощенное основание носа с искривлением носовой перегородки, нос может быть длинным, тонким, с заостренным или крючковидным кончиком. Верхняя челюсть у таких больных часто недоразвита, что вместе с высоким лбом формирует уплощенный лицевой профиль. Нёбо часто сужено, с высоким сводом и иногда с расщелиной. Аномалии зубов включают сверхкомплектные зубы, гипоплазию эмали и нарушения образования дентина, приводящие к истончению и сужению корней. Деформации конечностей включают частичную мягкотканную синдактилию между II и III пальцами кисти или стопы. Также наблюдаются брахидактилия, клинодактилия и раздвоение дистальных фаланг.
ДИАГНОСТИКА:
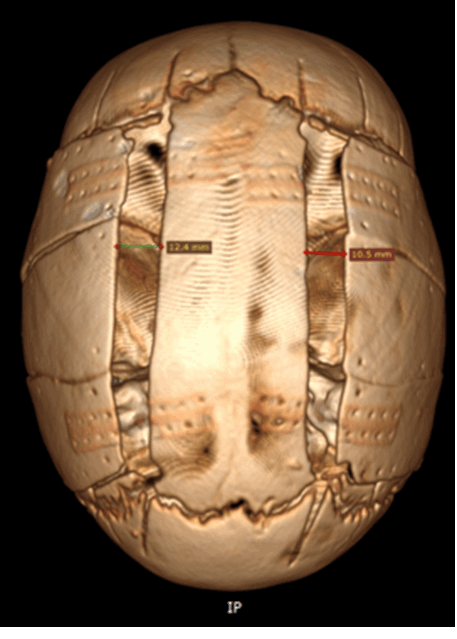
- МСКТ костей черепа и головного мозга с трехмерной реконструкцией наиболее информативный метод диагностики. Позволяет достоверно выявить преждевременно заросший шов, подтвердить, клинически выявленную, деформацию формы головы.
- УЗИ костей черепа так же позволяет выявить преждевременное заращение швов черепа.
Показания к хирургическому лечению:
- Краниосиностоз
- Повышение внутричерепного давления
- Деформация челюстно-лицевой системы
Противопоказания к оперативному лечению:
Воспалительный процесс любой локализации в стадии обострения, неполной ремиссии
Анемия средней и тяжелой степени.
Тяжелое общесоматическое состояние
Обострение хронических заболеваний.
Тяжелое общесоматическое состояние пациента (Кома 2-3).
Окончательное решение о наличии или отсутствии показаний к оперативному лечению принимает врач-нейрохирург в рамках очной или заочной консультации.
Особенности послеоперационного периода:
Послеоперационный период и реабилитационные мероприятия носят сугубо индивидуальный характер.
Количество койко-дней: 6-21.
Необходимость в повторной явке: В послеоперационном периоде пациенту необходим динамически МРТ-контроль, с последующей консультацией нейрохирурга.
Деформации черепа/Краниосиностозы
Не совсем правильно называть деформацию черепа краниостенозом, т.к. краниостеноз – конечный результат патологического процесса.
Частота встречаемости в РФ деформаций черепа связанных с краниосиностозом у детей приходится 1:2000 новорожденных.
Анатомия и физиология швов.
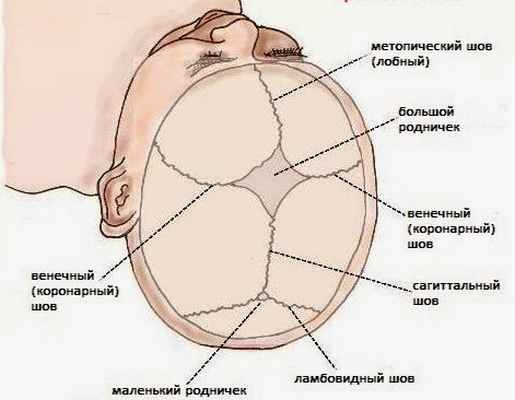
Рис.№1 Анатомия швов.
Кости свода черепа человека соединяются между собой с помощью эластичных швов.
Швы имеют большое значение в биомеханики родов, благодаря им происходит смещение, временная деформация черепной коробки при прохождении по родовым путям ребенка с целью минимизации травматического повреждения головного мозга. Т.е. швы придают мобильность ригидной (плотной) структуре черепа.
Помимо этой функции, швы также препятствуют преждевременному слиянию костей черепа между собой, тем самом, создавая оптимальные условия к росту черепа.
Известно, что за первые 6 месяцев жизни ребенка, размер черепной коробки увеличивается вдвое.
Преждевременное слияние частей черепа между собой приводит к развитию краниосиностоза, деформации черепа.
Причины (этиология) возникновения деформации черепа:
1. Внутриутробные
2.Генетические (встречается в 20% случаев).
3. Пороки развития черепа (энцефалоцеле, микроцефалия).
4.Близкородственные браки.
5. Отягощенный акушерский анамнез, наличие самопроизвольных абортов, ОРВИ/другие вирусные инфекции перенесенные на 1-2-м месяце беременности и др. Курение, алкоголь также являются провоцирующими факторами к формированию патологического процесса.
6. Эндокринно-обменная теория – гипертериоз. Именно щитовидная железа оказывает наибольшее влияние на развитие и формирование костных швов.
Клинические проявления.
1.Синдром повышенного внутричерепного давления (ВЧД) - головные боли, частые обильные срыгивания, рвота, выраженное беспокойство в поведении ребенка. Такие проявления встречаются в большинстве случаев 60-70%.
2.Нарушение зрения, косоглазие, слезотечение, экзофтальм (выпячивание глазного яблока вперед), атрофия зрительного нерва (что приводит к слепоте ребенка).
3.Отставание в психомоторном развитии – позднее сверстников начинаю самостоятельно сидеть, ходить, говорить, в более старшем возрасте
возрасте низкая учебная успеваемость. Отставание в психомоторном развитии, чаще у детей проявляется в дошкольном и школьном периодах.
4. Косметический дефект. Психологические нарушения адаптации в обществе. С возрастом (чаще в подростковом периоде) именно эта проблема будет основополагающей, т.к. у ребенка с возрастом формируется психологическая травма.
5.Судороги – развитие эпилепсии.
«Золотым» стандартом в исследовании любой деформации черепа у детей любой возрастной категории, является спиральная компьютерная томография (СКТ).
Боязнь родителей «облучить» ребенка уходит в прошлое. Прогрессирующая деформация черепа – краниосиностоз/краниостеноз крайне отрицательно влияет на развитие головного мозга растущего ребенка и является страшнее всех предрассудков.
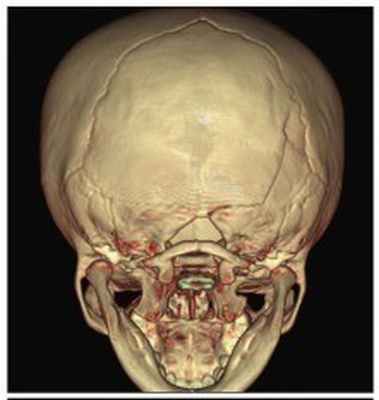
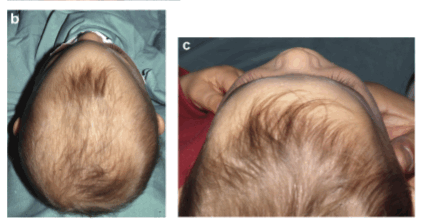
Рис. №2 а) – вариант нормы
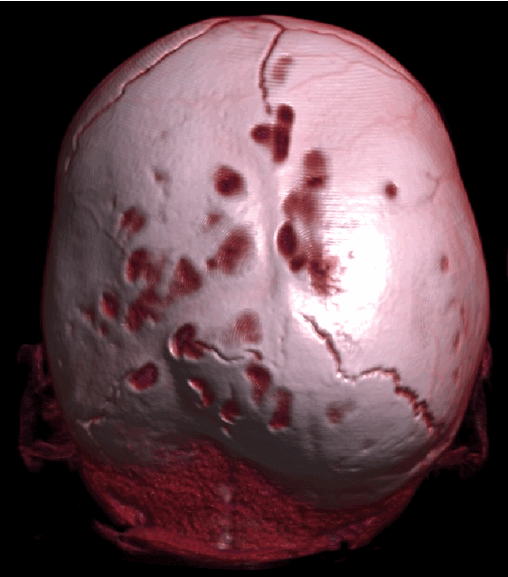
б) – деформация черепа вызванная синостозом (слиянием) лямбдовидного шва слева, указателем показаны множественные пальцевые вдавления (истончение костной ткани, вплоть до появление «дыр»).
Лечение прогрессирующей деформации черепа вызванной на фоне краниосиностоза/краниостеноза (преждевременного сращения костного шва/швов) только хирургическое.
Медикаментозная терапия и лечение ортезами (ортопедические падушки, шлемы) при прогрессирующей деформации черепа (краниосиностозе, краниостенозе) не эффективно и может усугубить состояние ребенка.
Самый оптимальный возраст для хирургического лечения, является возраст ребенка с 6 мес. до 1 года.
Суть операции заключается не только в устранении косметического дефекта, но и в увеличении объема полости черепной коробки – именно последний факт обеспечивает благоприятные условия для развития головного мозга.
Не все деформации черепа подвергаются хирургической коррекции. Тактика лечения определяется нейрохирургом совместно с неврологом.
Для реконструктивных операций в нашей клинике используются современные саморассасывающиеся (биодеградируемые) имплантаты, так и артифициальные титановые импланты зарубежных производителей – Inion, Stryker, DePuy…
Операции детям Российской Федерации проводятся в рамках оказания высокотехнологической медицинской помощи за счет средств обязательного медицинского страхования.
Примеры хирургической коррекции деформации черепа.
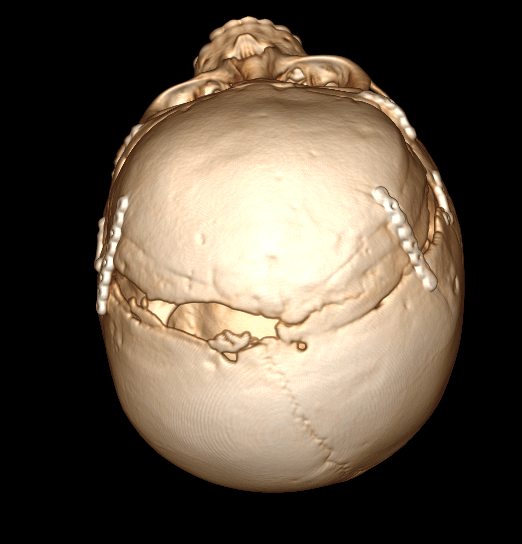
На сегодняшний день, во всем мире (и наша клиника, согласна с этим мнением) прибегают к открытым реконструктивным методам хирургической коррекции краниостеноза, тем самом уходя от применения эндоскопического лечения деформаций черепа. При эндоскопической коррекции до 80% случаев приходится проводить реоперации, т.к. устраняется только косметический дефект черепа не увеличивая объем полости черепа (см.выше)
Деформация черепа, вызванная преждевременным слиянием (стенозом) сагиттального шва - Скафоцефалия.
Скафоцефалия - стеноз сагиттального шва (от греч. scapho– «ладья»). Занимает первое место по числу встречаемости (0,2-1 случай из 1000 новорожденных). См. Рис. №3
При таком поражении имеет место сужение черепа по бокам за счет уменьшения роста теменных и височных костей и компенсаторный рост в переднезаднем отделах.
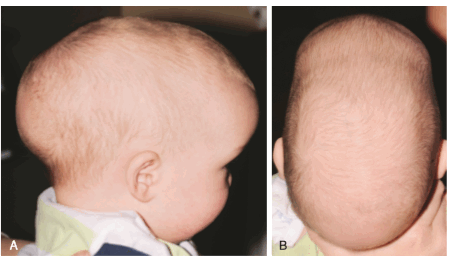
а) – форма черепа ребенка при скафоцефалии
б) - эндоскопическая коррекция (СКТ снимки)
в) – открытая реконструкция костей свода чрепа (СКТ снимки)
Деформация черепа, вызванная преждевременным слиянием (стенозом) метопического шва – Тригоноцефалия.
Тригоноцефалия – стеноз метопического шва (метопический шов – разделяет две лобные кости между собой). Происходит названием от греч. trigonos – треугольный. Характеризуется клиновидной деформацией лобных костей. Всегда сопровождается деформацией глазниц.
Физиологическое закрытие метопического шва происходит в возрасте от 3-9 месяцев до 2 лет.
Рис. №4 Тригоноцефалия
б) спиральная компьютерная томография (СКТ) при тригоноцефалии
в) СКТ снимки через 1 сутки после операции - открытой реконструкции
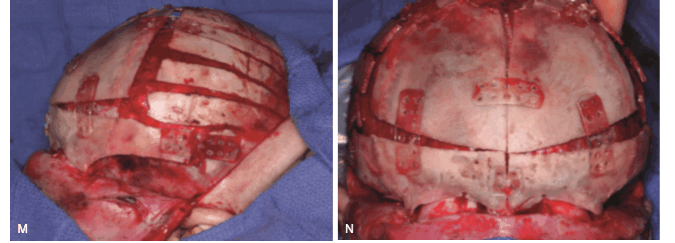
г) интраоперационные снимки
д) сравнение до и после операции.
Деформация черепа, вызванная преждевременным односторонним слиянием (стенозом) коронарного шва – Плагиоцефалия.
Плагиоцефалия (от греч. Рlagio – косой + kephale – голова) – косая асимметрия черепа. Может быть лобной и затылочной.
Синостозна лобная плагиоцефалия или гемикоронарный синостоз (ГКС) – связан с односторонним стенозов коронарного шва, который привод к заращению лобно-клиновидного и лобно-решетчатого швов на той же стороне. Всегда сопровождается деформацией костей лицевого скелета.
Рис. №5 Плагиоцефалия
а), б) - СКТ снимки плагиоцефалии
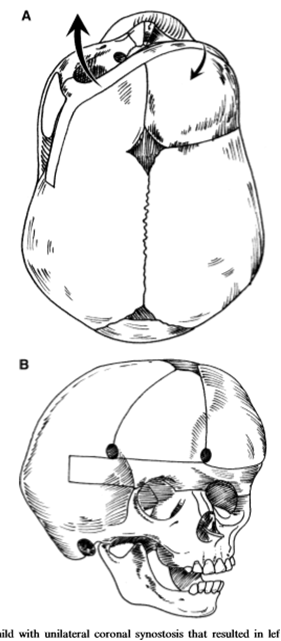
в), г) - схема хирургического лечения. Выдвижение вперед асимметричной части черепа и ее фиксация – фронто-орбитальная реконструкция.
Брахиоцефалия – (от греч. brachy – короткий) возникает в следствии синостозирования коронарных швов с 2-х сторон. Характеризируется укорочением в передне-задних отделах и компенсаторным удлинением вверх в височных костях.
Метод лечения, также является хирургическим. При данной форме краниосиностоза используются дистракторы (выдвижная система), с помощью которых в течение 20-30 дней (ежедневно на 0,1-0,2мм) увеличивается объем черепа. По окончанию срока и удовлетворительному результату, данная система удаляется.
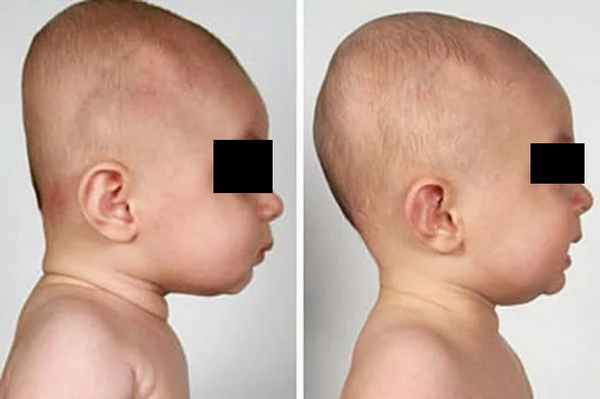
Рис. № 6 Брахиоцефалия
а) - до и после операции
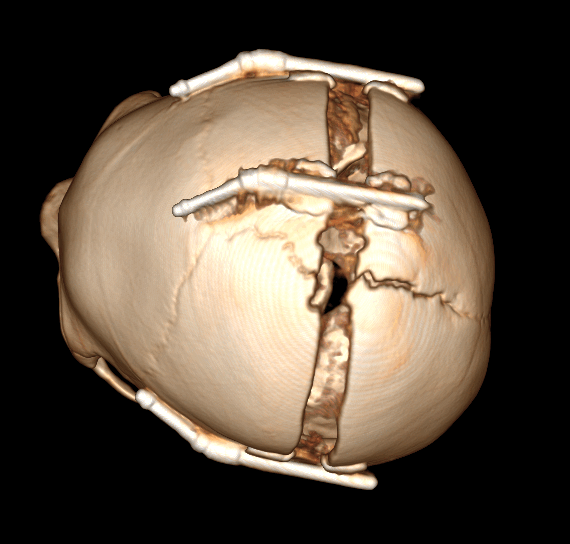
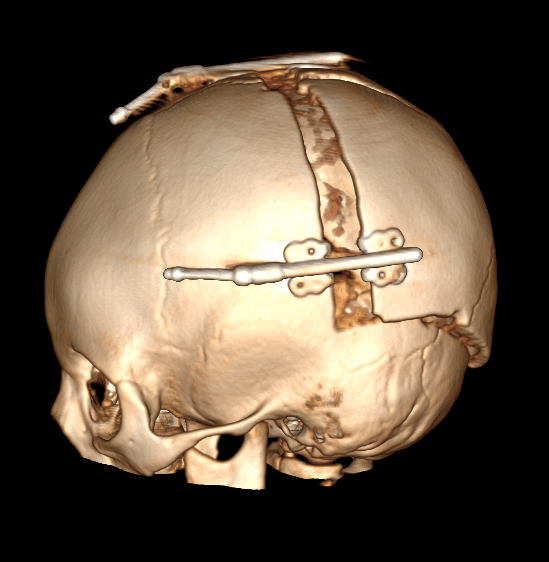
б), в) - СКТ снимки после установки дистракторов.
Синдромальные краниосиностозы (Аперта, Крузона, Пфейффера и т.д.), требуют командной работы (генетика, челюстно-лицевого хирурга, окулиста), и как правило положительный результат достигается путем этапного хирургического лечения.
Согласие на обработку персональных данных
Адрес: 344015, г. Ростов-на-Дону, ул. 339-й Стрелковой Дивизии, 14
© 2022 ГБУ РО «Областная детская клиническая больница». ВСЕ ПРАВА ЗАЩИЩЕНЫ.
"Мы обратились к нейрохирургам, чтобы получить второе мнение, и теперь мы рады, что прооперировали нашего сына здесь".
Семья из Доминиканской Республики рассказывает, как прошла операция по исправлению краниосиностоза у их ребенка.

«Все началось тогда, когда Луис Карлос только родился, во время первого осмотра головы педиатр заметил, что у нашего сына закрылись роднички», - так Патриция рассказывает о моменте, когда лечащий врач впервые сказал им, что в черепе ее ребенка может быть деформация. «Наш врач предупредил нас, что это может быть краниосиностоз, и сказал, что мы будем наблюдать и измерять голову мальчика. Две недели спустя он увидел, что сагиттальные швы закрылись, поэтому ребёнку сделали компьютерную томографию, результат которой подтвердил диагноз "краниосиностоз"».
Врачи объяснили Патриции и ее мужу, что решение проблемы -- это нейрохирургическая операция и что ее нужно делать как можно быстрее. «В тот момент мы начали задумываться о том, чтобы уехать из нашей страны и запросить второе мнение, так как в Доминиканской Республике хотя и есть хорошие нейрохирурги, но почти нет педиатров», - рассказывает мама мальчика. В Санто-Доминго семье посоветовали поехать в США или Европу, если необходимо будет оперировать ребенка.
Тогда они связались с Госпиталем Сант Жоан де Деу Барселона, чтобы запросить второе мнение у нейрохирургов госпиталя.
«Они связались с нами, потому что я прооперировал пациента из Центральной Америки с похожим случаем», - говорит Хосе Инохоса, заведующий Отделением детской нейрохирургии. «Мнение специалистов нашего госпиталя отличалось от других мнения других специалистов касаемо возраста и используемой техники, так как мы используем различные техники и оперируем детей с четырех месяцев».
В случае краниосиностоза, очень важно как можно быстрее провести операцию.
Нейрохирург поясняет, что ребенку поставили правильный диагноз в стране происхождения. «Краниосиностоз - это раннее закрытие черепных швов, которое вызывает деформацию черепа и повышение давления в головном мозге. Поэтому хорошо, что они обнаружили краниосиностоз у ребёнка немедленно, так как дети, страдающие им, должны быть прооперированы как можно скорее", - подводит итог Хинохоса. Хирургическая техника, применяемая к этим пациентам, зависит от каждого госпиталя и типа имеющейся у ребенка деформации черепа.
В Госпитале Сант Жоан де Деу Барселона детей с этим пороком развития оперируют предпочтительно в возрасте от трех до пяти месяцев. Проблема обычно решается эндоскопией - малоинвазивным методом с отличными результатами. В данном случае, поскольку это был ребенок старше шести месяцев, был выбран другой метод, менее распространенный, который представляет собой вариацию по сравнению с методами, используемыми несколько лет назад.
«Речь идет о технике H и Ω (Омега), с помощью которой мы можем вернуть черепу нормальные пропорции, решить проблему повышенного черепного давления и, что очень важно, реконструировать череп так, чтобы не было дефектов после операции», - поясняет заведующий нейрохирургическим отделением госпиталя Хосе Инохоса. Раннее вмешательство у этих пациентов также позволяет получить хороший результат с косметической точки зрения.
«Благодаря поддержке блестящей команды экспертов-анестезиологов и первоклассного педиатрического отделения интенсивной терапии, в нашем центре можно проводить нейрохирургические операции в возрасте около шести месяцев. По факту, это сложная операция, которую мы проводим относительно регулярно и с хорошими результатами», - заключает Хосе Инохоса.
Данные подтверждают эту информацию: только в 2020 году было выполнено более 50 хирургических вмешательств при краниосиностозе с вероятностью успеха 99%, с относительно быстрым периодом восстановления и практически без каких-либо осложнений. Маленький Луис Карлос является примером, так как он был выписан всего через пять дней после операции, благодаря его быстрому восстановлению.
«В течение нескольких месяцев мы спрашивали у детских нейрохирургов второе мнение, и мы счастливы, что наконец прооперировали нашего сына здесь», - говорит мать Луиса Карлоса.
«С помощью рисунков врач объяснил нам, какая операция предстоит Луису, и успокоил нас, потому что, когда врачи говорят, что будут оперировать голову вашего ребенка, это сильно тревожит. Но нам предоставили много информации, и мы очень довольны», - резюмирует Патрисия, добавляя, что поездка для проведения нейрохирургической операции во время пандемии была нелегкой, но они получили необходимую поддержку от команды Отдела по работе с иностранными пациентами. «Благодаря гостеприимству и индивидуальному подходу, мы чувствовали себя как дома с момента прибытия в Барселону и до отъезда».
Краниосиностоз
Краниосиностоз – это заболевание, основным симптомом которого является деформация мозгового отдела черепа, возникающая вследствие преждевременного зарастания костных швов. Клиника включает в себя деформации черепа, симптомы внутричерепной гипертензии, патологию зрительного нерва, отставание в психическом развитии. Редко заболевание сопровождается аномалиями костей лицевого черепа. Диагностика заключается в оценке степени зарастания черепных швов и определении костных дефектов путем физикального обследования, рентгенографии, КТ и МРТ. Основное лечение – ранняя хирургическая коррекция формы костей черепа.
Общие сведения
Краниосиностоз – это патологическое состояние в педиатрии, возникающее на фоне раннего зарастания черепных швов, характеризующееся деформацией черепной коробки и нарушением развития тканей головного мозга. В среднем распространенность разных форм заболевания в станах СНГ составляет 0,03-3,5% от всех новорожденных. Мужской пол более склонен к развитию данной патологии. Наиболее распространенный вариант – моносиностоз. Чаще всего наблюдается преждевременное зарастание сагиттального шва (скафоцефалия) – 50-65% от всех краниосиностозов. Самой редкой и прогностически неблагоприятной является синдромальная форма, при которой имеется высокий риск летального исхода на первом году жизни ребенка. При своевременной диагностике и адекватном лечении в первые 6-9 месяцев жизни дальнейшее развитие пациента проходит без отклонений.
Причины краниосиностоза
Точная этиология краниосиностоза не установлена. Согласно выдвинутым теориям, данное заболевание может развиваться в результате внутриутробного нарушения гормонального фона ребенка, перинатальных травм и сдавливания костей черепа в полости матки. Также данная патология возникает при наследственных патологиях – синдроме Апера, синдроме Крузона и синдроме Пфайффера. Доподлинно известна одна из ведущих причин развития краниосиностоза – аномалия гена, отвечающего за образование рецепторов фактора роста фибробластов (FGFR типы I, II и III).
Патогенетически краниосиностоз обусловлен преждевременным синостозированием одного или сразу нескольких черепных швов: коронарного, сагиттального, лямбдовидного или метопического. На фоне этого, согласно закону Вирхова, возникает компенсаторный рост костной ткани в перпендикулярном направлении, из-за чего формируется деформация черепа. Полисиностоз (а зачастую – и моносиностоз) часто сопровождается внутричерепной гипертензией, которая может проявляться неврологическими нарушениями вследствие сдавливания коры головного мозга, венозным застоем глазного дна, отеком диска зрительного нерва, а при длительном течении – полной атрофией зрительного нерва и потерей зрения.
Классификация краниосиностоза
Краниосиностоз, согласно этиологическим факторам, разделяют на две группы:
- Синдромальный. В данном случае патология сочетается с другими врожденными пороками. Сюда относятся сцепленные с Х-хромосомой, моногенные, хромосомные и другие краниосиностозы. Например – комбинация синостоза с дисплазией костей лицевого черепа, синдром Смита-Лемли-Опица или рото-пальце-лицевой синдром.
- Несиндромальный. Это изолированная форма, которая возникает самостоятельно и не имеет сопутствующих заболеваний.
В зависимости от количества заросших черепных швов выделяют:
- Моносиностоз. Характеризуется поражением только 1 шва. В случае с коронарным и лямбдовидным швом зарастание может быть одно- или двухсторонним. Наиболее распространенная форма.
- Полисиностоз. В патологический процесс втягиваются 2-3 шва.
- Пансиностоз. При этой форме наблюдается сращивание всех костных швов черепа ребенка. Встречается крайне редко.
Симптомы краниосиностоза
Клинически краниосиностоз проявляется с момента рождения ребенка. Для всех форм характерны плагиоцефалия и раннее закрытие большого родничка (в норме это происходит в 12-18 месяцев). Только при полисиностозе или сопутствующей гидроцефалии он может оставаться открытым до 3-х летнего возраста. Также при краниосиностозах зачастую наблюдается повышение внутричерепного давления, которое может проявляться неврологическими нарушениями: беспокойством, интенсивным плачем, тошнотой и рвотой, нарушением сна, снижением аппетита, позитивным симптомом Грефе, судорогами.
Каждая из форм заболевания имеет характерные клинические особенности. Краниосиностоз стреловидного шва (скафоцефалия или ладьевидный череп) характеризуется увеличением переднезаднего размера головы ребенка при недостаточности ее ширины. Визуально определяется вытягивание черепа, «вдавливание» височных областей, «нависание» лба и затылочной части, сужение лица и приобретение им овальной формы. Пальпаторно над местом прохождения стреловидного шва выявляется костный гребень. В раннем возрасте возможна задержка психического развития.
Зарастание лямбдовидного шва чаще всего носит односторонний характер и проявляется уплощением затылочной области. Является трудно диагностируемой формой, поскольку плагиоцефалия практически незаметна под волосами, а неврологические нарушения минимальны. При взрослении пациента динамика заболевания практически отсутствует.
Коронарный или венечный краниосиностоз может быть как одно-, так и двухсторонним. Зарастание только одной половины шва сопровождается типичной деформацией черепа ребенка – уплощением лобной кости и верхней части глазницы с пораженной стороны. При этом противоположная половина компенсаторно «нависает». Со временем развиваются искривление носа в противоположную сторону, уплощение скулы, нарушение прикуса и косоглазие. Двухсторонний коронарный краниосиностоз проявляется широким, плоским и высоким лбом с уплощенными глазничными краями лобной кости, редко – башенной деформацией черепа (акроцефалией). Неврологические нарушения неспецифичны и аналогичны другим формам.
Нетопический краниосиностоз или тригоноцефалия характеризуется развитием треугольного лба с костным килем, проходящим от глабеллы до большого родничка. Также наблюдается гипотелоризм – смещение глазниц кзади с уменьшением межглазничного промежутка. Со временем происходит некоторое сглаживание костного гребня и нормализация формы лба. В половине случаев возникают нарушения зрения и отставание в психическом развитии.
Синдромальный краниосиностоз является самой редкой и тяжелой формой. Помимо плагиоцефалии отмечается дисплазия костей лицевой части черепа, из-за чего возникают дыхательная недостаточность, нарушение приема пищи и патология зрения. Характеризуется синостозом венечного шва и, как результатом – брахицефалической формой головы ребенка. Также возникают гипоплазии костей верхней челюсти, выпячивание глазных яблок из орбит, гипертелоризм. Часто наблюдается значительное расширение родничка и расхождение стреловидного шва. Без лечения у детей развивается выраженное отставание в психическом развитии, зачастую они погибают на протяжении первых 12 месяцев жизни от ОРВИ, осложнившихся пневмонией.
Диагностика краниосиностоза
Диагностика краниосиностоза базируется на физикальном осмотре и инструментальных методах исследования. Анамнез нередко малоинформативен, но его данные позволяют педиатру проследить динамику клинической симптоматики, если таковая имеет место. Важным моментом становится визуальный осмотр ребенка, который дает возможность обнаружить характерные деформации черепа, аномалии костей и т. д. Лабораторные анализы специфических изменений не выявляют и могут использоваться с целью определения генетической патологии или диагностики осложнений.
Обязательными являются инструментальные методы, позволяющие визуализировать костные деформации и оценить степень поражения тканей головного мозга. Сюда относятся нейросонография, рентгенография, компьютерная и магнитно-резонансная томография. Нейросонография используется с целью оценить состояние тканей головного мозга и размеры желудочков, выявить внутричерепную гипертензию. На рентгенограмме удается определить нарушения структуры костей, окостенение черепных швов, а при повышенном внутричерепном давлении – усиление пальцевых вдавлений. КТ и МРТ применяются для получения более информативных результатов. При подозрении на поражение зрительной системы проводится офтальмоскопия, позволяющая обнаружить поражение диска зрительного нерва. Рекомендованы консультации нейрохирурга и офтальмолога.
Дифференциальная диагностика краниосиностоза осуществляется с позиционной плагиоцефалией, родовой травмой новорожденных (кефалогематомой, подапоневротическим кровоизлиянием, переломом костей черепа), кистами головного мозга, рахитом и микроцефалией.
Лечение краниосиностоза
Основное лечение краниосиностоза – хирургическая коррекция костной деформации черепа. Оптимальное время для проведения оперативного вмешательства – первые 6-9 месяцев жизни ребенка. Данные сроки обусловлены тем, что в этом периоде наблюдается наиболее интенсивное развитие тканей головного мозга, которому может препятствовать деформация черепной коробки. Кроме того, кости черепа в этом возрасте быстро восстанавливают свою структуру без развития осложнений. Объем и техника операции зависят от формы краниосиностоза и сопутствующих патологий. В 2-3-х летнем возрасте коррекция проводится исключительно с целью ликвидировать косметический дефект. Помимо хирургического лечения осуществляется изменение рациона ребенка в соответствии с возрастными требованиями. При развитии интеркуррентных заболеваний показана медикаментозная терапия.
Прогноз и профилактика краниосиностоза
Прогноз для детей с краниосиностозом напрямую зависит от формы заболевания, своевременности диагностики и эффективности оперативного вмешательства. При качественном проведении лечебных мероприятий исход заболевания, как правило, благоприятный. Прогностически неблагоприятной принято считать синдромальную форму краниосиностоза.
Специфической профилактики для данной патологии не существует. Неспецифические меры подразумевают медико-генетическую консультацию семьи и планирование беременности, охрану здоровья женщины при вынашивании ребенка, рациональное питание, отказ от вредных привычек и исключение всех потенциальных этиологических факторов развития краниосиностоза.
Краниосиностоз
Преждевременное слияние швов вызвает характерную деформацию черепа из-за снижения роста в направлении, перпендикулярном закрытому шву. Это наблюдается у 1 из 2500 живорожденных детей. Существует несколько типов, в зависимости от того, какие швы сливаются.
Черепные швы
Сагитальный краниосиностоз
Сагитальный краниосиностоз является наиболее распространенным типом и приводит к узкому и длинному черепу (долихоцефалия). Большинство случаев являются изолированными и спорадическими с риском повтора передачи вируса будущему потомству < 3%. Неспособность к обучению может наблюдаться у 40-50% больных. При сагиттальном краниосиностозе задействовано несколько генов, однако специфическое генетическое тестирование в настоящее время не рекомендуется, если только не присутствуют другие врожденные аномалии.
Коронарный краниосиностоз
Венечный краниосиностоз является вторым наиболее распространенным типом и может быть двусторонним, вызывая формирование короткого широкого черепа (брахицефалия), или односторонним, в результате чего формируется диагональная деформация черепа (плагиоцефалия). Истинная плагиоцефалия (т.е. вызванная краниосиностозом) часто приводит к асимметричным орбитам и должна быть дифференцирована от позиционной плагиоцефалии, которая возникает из-за кривошеи Врожденная кривошея Аномалии шеи и спины могут быть вызваны повреждением мягких тканей или костей либо аномалиями позвоночника. (См.также Введение во врожденные черепно-лицевые и скелетно-мышечные расстройства. Прочитайте дополнительные сведения или укладывания младенца преимущественно на одну сторону и не приводит к асимметричным орбитам. При позиционной плагиоцефалии задняя часть черепа с одной стороны сплюснута, наблюдается фронтальная выпуклость на той же стороне, ухо на сплюснутой стороне может быть выдвинуто вперед, но орбиты остаются симметричными. Около 25% случаев венечных краниосиностозов являются синдромными и возникают из-за мутаций одного гена или хромосомных дефектов. У пациентов с изолированным несиндромальным коронарным краниосиностозом были выявлены мутации в нескольких генах. В частности, у 32% пациентов с двусторонним венечным краниосиностозом и у 10% пациентов с односторонним венечным краниосиностозом наблюдаются мутации гена TCF12 ( 1 Венечный краниосиностоз, ссылки Краниосиностоз является преждевременным слиянием одного или более черепных швов. (См.также Введение в врожденные черепно-лицевые и скелетно-мышечные расстройства (Introduction to Congenital. Прочитайте дополнительные сведенияВенечный краниосиностоз обычно ассоциируется с лицевыми и экстракраниальными аномалиями в контексте синдромов Крузона, Мюнке, Пфайффера, Сетре-Чотзена, Карпентерра или Аперта.
Венечный краниосиностоз, ссылки
1. Sharma VP, Fenwick AL, Brockop MS, et al: Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet 45(3):304–307, 2013. doi: 10.1038/ng.2531. Epub 2013 Jan 27. Clarification and additional information. Nat Genet 45(10):1261, 2013.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: