КТ, МРТ при гемифациальной микросомии
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Cинонимы: синдром первой и второй жаберных дуг, синдром Гольденхара – окуло-ауриколо-вертебральная дисплазия, краниофациальная микросомия, отомандибулярный дизостоз и латеральная фациальная дисплазия - является редким наследственным заболеванием, характеризующимся значительным числом аномалий, которые возникают вследствие нарушения развития первой и второй жаберных дуг первого глоточного кармана, первой жаберной щели и зачатков височной кости. Среди врожденных пороков развития черепно-челюстно-лицевой области гемифациальная микросомия занимает второе место по частоте встречаемости после врожденных расщелин верхней губы и нёба. Частота этого синдрома колеблется в пределах 1:3500-5600 новорожденных, он присутствует у 1 из 1000 у детей с врожденной глухотой. Распределение по половому признаку (мужчины и женщины) составляет примерно 3:2.
Этиология и тип наследования изучены недостаточно. Неблагоприятный акушерско-гинекологический анамнез матери (предшествующие аборты, сахарный диабет, избыточный вес) и тератогенные факторы на ранних сроках беременности являются отягощающими факторами риска рождения больного ребенка. Вероятность повторного рождения больного ребенка, как и вероятность рождения больного ребенка у носителя патологии, ориентировочно равна 2%.
Клинически для гемифациальной микросомии характерны: недоразвитие тела и ветви нижней челюсти, гипоплазия скуловой кости и дуги, недоразвитие структур ВНЧС; аплазия ветви нижней челюсти и ВНЧС; нарушение размеров и положения глазницы; гипоплазия, аплазия ушной раковины, атрезия слухового прохода, поражение лицевого нерва, гипоплазия мимических мышц; дефицит мягких тканей; макростомия; предушные придатки и свищи; иногда сочетание с врожденной расщелиной губы и неба, эпибульбарным дермоидом, аномалией прикуса, адентией, нарушением структуры эмали и формы зубов, пороками развития опорно-двигательного аппарата, органов зрения и нервной системы, а также аномалиями мочевыделительной системы и желудочно-кишечного тракта.
Cклеродермия
Это прогрессирующее системное заболевание, в основе которого лежит воспалительное поражение мелких сосудов всего организма, с последующими фиброзно-склеротическими изменениями кожи, опорно-двигательного аппарата и внутренних органов.
При очаговой склеродермии наблюдается ограниченное уплотнение кожи, но могут вовлекаться подкожные ткани и кости. Выделяют две основные формы очаговой склеродермии – бляшечную (морфея) и линейную. В первом случае поражение кожи имеет вид уплотнений округлой формы («Бляшки»), с лиловым ободком по периферии в дебюте болезни. Эти очаги, единичные или множественные, могут появляться как на туловище, так и на лице и конечностях. При линейной форме очаговой склеродермии участки поражения имеют вид полос уплотнения кожи, часто с вовлечением подлежащих мышц и костей, и локализуются, главным образом, на конечностях и лице. Эта форма очаговой склеродермии в случае развития в детском и подростковом возрасте, может приводить к ограничению движений (мышечные и суставные контрактуры) и нарушениям развития пораженных участков. Внутренние органы при очаговой склеродермии не страдают.
Системная склеродермия (ССД) – форма склеродермии, при которой помимо уплотнений кожи развиваются разнообразные поражения суставов, внутренних органов (сердца, легких, желудочно-кишечного тракта, почек). В редких случаях наблюдается поражение только внутренних органов, без изменений кожи. Женщины заболевают в 3-5 раза чаще, чем мужчины. ССД подразделяется на лимитированную и диффузную форму, которые различаются по распространенности и выраженности поражения кожи и внутренних органов.
Липодистрофия
Общее или локальное поражение подкожной клетчатки с уменьшением (атрофическая форма) или увеличением (гипертрофическая форма) объема жировой ткани. Л. могут быть генерализованными или сегментарными. К липодистрофиям относят следующие патологические состояния: врожденную генерализованную Л., гипермускулярную Л., прогрессирующую сегментарную Л., или болезнь Барракера — Симонса, постинъекционную Л., Липоматоз болезненный (болезнь Деркума).
Прогрессирующая сегментарная липодистрофия (синонимы липоатрофия, болезнь Барракера-Симона) — это Относительно редкое заболевание, проявляющееся атрофией подкожной жировой клетчатки лица, шеи, плечевого пояса, грудной клетки при нормальном или избыточном отложении жира в нижней половине тела. Встречается редко, поражает преимущественно женщин. Этиология неясна. Внутренние органы не поражаются, функция их не нарушена. Больные обращаются к врачу из-за своей внешности, иногда жалуются на слабость, раздражительность. Жизненный прогноз благоприятен. Специфичного лечения нет, применяют средства, укрепляющие нервную систему, и витамины.
Микрогения
Это аномалия развития лица, характеризующееся гипоплазией (недоразвитием) нижней челюсти.
В основе деформации лежит нарушение роста и развития нижней челюсти в результате травмы или патологического процесса в области сустава и ветви челюсти, перенесенных во время родов, в детском возрасте при незаконченном росте лицевого скелета, а также вследствие врожденных нарушений во время развития нижне- и верхнечелюстного отростков головной части зародыша. При односторонней микрогении лицо асимметрично за счет укорочения ветви и горизонтальной части тела челюсти, что приводит к смещению подбородка в сторону укорочения. Пораженная сторона выглядит более выпуклой, здоровая сторона уплощена, тело челюсти удлинено и деформировано. Двусторонняя (симметричная) микрогения характеризуется укорочением тела и ветвей челюсти, что сопровождается смещением подбородочного отдела кзади и его «скошенностью». При этом верхняя челюсть выступает вперед, придавая лицу так называемое «птичье выражение». Деформация нижней челюсти может быть выражена в различной степени: от незначительной асимметрии или уплощения подбородочного отдела с умеренным нарушением прикуса до тяжелых, обезображивающих деформаций. При выраженном нарушении прикуса развиваются функциональные расстройства из-за затруднения акта откусывания и пережевывания пищи.
Микрогения довольно часто сочетается с анкилозом височно-челюстного сустава.
КТ, МРТ при гемифациальной микросомии
а) Терминология:
1. Аббревиатура:
• Гемифациальная микросомия (ГФМ)
2. Синонимы:
• Окуло-аурикуло-вертебральная дисплазия, синдром Гольденхара, фацио-аурикуло-вертебральная последовательность
3. Определение:
• Дефект производных 1-й и 2-й глоточной дуги ± клеток нервного гребня:
о Аномалии внутреннего и среднего уха
о Типичная односторонняя гипоплазия нижней челюсти и ВНЧС
б) Визуализация:
1. Общая характеристика:
• Лучший диагностический критерий:
о Односторонняя гипоплазия нижней челюсти, отсутствие скуловой дуги, атрезия/стеноз ИСК
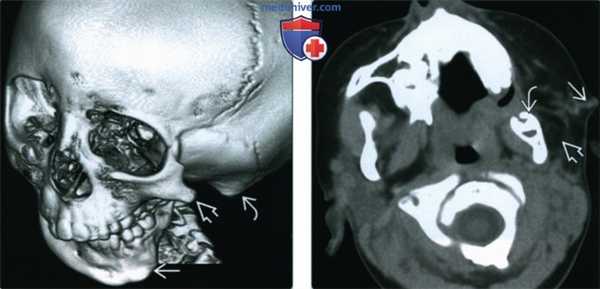
(Слева) На 3D КТ у семилетней девочки с гемифациальной микросомией (ГФМ) определяется уменьшение нижней челюсти слева, гипоплазия скуловой дуги, недоразвитие сосцевидного отростка и атрезия наружною слуховою канала.
(Справа) При аксиальной КТ без КУ у четырехлетней девочки с ГФМ определяется уменьшение дуги нижней челюсти слева, визуализируется преаурикулярный свищевой ход, а также «метка» на коже. Жевательные мышцы слева недоразвиты; жевательная мышца и околоушная железа не визуализируются в ожидаемом месте.
2. КТ при гемифациальной микроскомии:
• КТ в костном окне:
о Микрогнатия:
- Обычно односторонняя, реже двухсторонняя или асимметричная; гипо- или аплазия ВНЧС
о Гипоплазия/отсутствие скуловой дуги
о Гипоплазия жевательных мышц и мышц лица
о Гипо-/аплазия околоушной железы ± добавочная околоушная железа
о Височные кости: врожденные аномалии наружного и среднего уха (СЕМЕМ):
- НСК: стеноз/атрезия, уменьшение/отсутствие тимпанической пластинки
- Сосцевидный отросток: ± вариабельное снижение пневматизации
- Пространство среднего уха: вариабельная гипоплазия
- Слуховые косточки: мальформация, ротация, слияние
- Овальное окно: ± стеноз или атрезия
- Канал ЧН VII (КЛН): норма или гипоплазия:
Тимпанический КЛН±расхождение/абберация
Сосцевидный КЛН вентральнее, иногда в ВНЧС
- Внутренне ухо: случайные мальформации
о Лицо: ± расщелины лица; ± других структур, например, глазниц
о Позвоночник: + аномалии слияния/сегментации
3. МРТ при гемифациальной микроскомии:
• Гипоплазия нижней челюсти и малых жевательных мышц
• Позвоночник: ± аномалии позвонков
• ЦНС: ± вентрикуломегалия, спорадически расщепление ствола мозга, гипоплазия мозжечка, цефалоцеле
• ЧН VII: норма, гипо- или аплазия
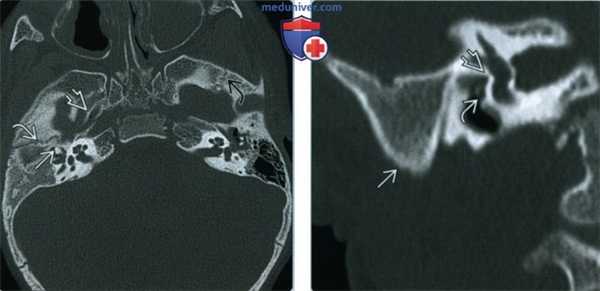
(Слева) При аксиальной КТ в костном окне у мальчика 23 месяцев с ГФМ определяется гипоплазия и снижение пневматизации полости среднего уха справа, дисморфия слуховых косточек. Евстахиева труба справа аномально расширена. Правый клиновидно-чешуйчатый шов В развернут кнаружи по сравнению с левым.
(Справа) На реформатированной корональной КТ в костном окне у этого же пациента отсутствует пневматизация сосцевидного отростка, определяется атрезия овального окна и аномальный ход тимпанического сегмента ЧН VII, проходящего через промонториум.
в) Дифференциальная диагностика гемифациальной микросомии:
1. Тератогенная эмбриопатия (например, диабет):
• Аномалии нижней челюсти и височных костей в зависимости от этиологии
2. Синдром Таунса-Брокса:
• Фенотипически имитирует синдром Гольденхара; СЕМЕМ; аномалии кисти, почек, анальной области
3. Бранхио-ото-ренальный синдром:
• Вариабельная асимметричная микрогнатия
• СЕМЕМ, расширение евстахиевых труб
• Коническое сужение базального завитка улитки; уменьшение, смещение среднего/апикального завитка
4. Синдром Тричера Коллинза:
• Двухсторонняя, обычно симметричная микрогнатия, гипоплазия скуловых дуг, СЕМЕМ
г) Патология:
1. Общая характеристика:
• Этиология:
о Частое заболевание, распространенность - один случай на 3500 новорожденных
о Генетические и внешние факторы
о Спорадическое, редко семейное заболевание (обычно аутосомно-доминантное; сниженная пенетрантность)
2. Стадирование, классификация гемифациальной микроскомии:
• Клиническая классификация OMENS (шкала тяжести):
о Асимметрия глазниц (О)
о Гипоплазия нижней челюсти (М)
о Деформация уха (Е)
о Дисфункция нервов(N)
о Дефицит мягких тканей (S)
д) Клинические особенности:
1. Проявления:
• Типичные признаки/симптомы:
о Асимметрия лица (100%): односторонняя микрогнатия ± гипоплазия верхней челюсти, расщелина, ± паралич ЧН VII
о Ухо (100%): микро-/анотия, преаурикулярные «метки» на коже, стеноз/атрезия ИСК, тугоухость (85%; кондуктивная > нейросенсорная)
о Глаза (72%): эпибульбарный липодермоид, колобома, микрофтальмия, нарушение зрения (28%)
о Деформация шейного отдела позвоночника о ЦНС: гидроцефалия, цефалоцеле, задержка умственного развития
• Другие признаки/симптомы:
о Трахеопищеводная фистула (ТПФ)/атрезия пищевода
о Аномалии сердца, легких, мочеполовых органов
2. Течение и прогноз:
• Вариабельная тяжесть, фенотипическая неоднородность
3. Лечение:
• Реконструкция нижней челюсти, слухопротезирование/операции на ухе
е) Дифференциальная диагностика. Следует учесть:
• ГФМ как причину асимметрии лица и аномалий наружного уха
• Аномалию/гипоплазию ЧН VII как причину ослабления лицевых мышц
ж) Список использованной литературы:
1. Beleza-Meireles A et al: Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet. 51(10):635-45, 2014
2. Gougoutas AJ et al: Hemifacial microsomia: clinical features and pictographic representations of the OMENS classification system. Plast ReconstrSurg. 120(7): 112e-120e, 2007
3. Kosaki R et al: Wide phenotypic variations within a family with SALL1 mutations: Isolated external ear abnormalities to Goldenhar syndrome. Am J Med Genet A. 143A(10):1087-90, 2007
4. Rahbar R et al: Craniofacial, temporal bone, and audiologic abnormalities in the spectrum of hemifacial microsomia. Arch Otolaryngol Head Neck Surg. 127(3):265-71, 2001
2. Синонимы:
• Окуло-аурикуло-вертебральная дисплазия, синдром Гольденхара, синдром 1-й и 2-й жаберной дуги, латеральная дисплазия лица, краниофациальная микросомия (КФМ)
3. Определение:
• Аномалия неясной природы с односторонним поражением уха, лица, нижней челюсти, зубов, глазницы
1. Общая характеристика:
• Лучший диагностический критерий:
о Недоразвитие мыщелка (маленький мыщелок—аплазия) и ипсилатеральной половины нижней челюсти
• Локализация:
о Производные 1-й и 2-й жаберных дуг: челюсти, мышцы, уши
о Пять важных краниофациальных проявлений ГФМ:
- Ухо: чаще всего поражающаяся мягкотканная структура
- Нижняя челюсть: наиболее часто поражающаяся костная структура
- Глазница
- Черепно-мозговые нервы: преимущественно ЧМН VII
- Мягкие ткани лица, включая жевательные мышцы
(Слева) На фотографии (выполненной при попытке улыбнуться) семилетней девочки с левосторонней ГФМ и параличом нижней части лица слева, обусловленным поражением ЧМН VII, определяется уменьшение размеров лица и ушной раковины елевой стороны.
(Справа) На КЛКТ (объемный рендеринг) у этой же пациентки лицевой скелет справа сформирован правильно. Вертикальный и горизонтальный размер левой половины нижней челюсти меньше, чем правой. Окклюзионная плоскость с левой стороны приподнята. Зубная срединная линия нижней челюсти смещена вправо, верхней челюсти - влево. (Слева) На панорамной реконструкции (КЛКТ) у этой же пациентки визуализируются правильно сформированные нижняя челюсть (в т.ч. мыщелок) и височная кость справа.
(Справа) На панорамной реформатированной КЛКТ у этой же пациентки слева визуализируется маленький мыщелковый отросток. Вертикальный размер ветви и тела нижней челюсти меньше по сравнению с противоположной стороной. (Слева) На КЛКТ (объемный рендеринг) у этой же пациентки левая половина нижней челюсти меньше по сравнению с противоположной стороной. Не полностью сформирована скуловая дуга, наружный слуховой канал отсутствует.
(Справа) На реформатированной косой сагиттальной КЛКТ визуализируется ВНЧС и проксимальная половина нижней челюсти. Обратите внимание на отсутствие суставного возвышения. Отсутствуют также структуры наружного и внутреннего уха, сосцевидный отросток уменьшен в размерах.
2. КТ при гемифациальной микросомии:
• Мыщелок уменьшен в размерах или отсутствует
• Суставная ямка сформирована пропорционально размерам и функции мыщелка
• Ипсилатеральная ветвь и тело нижней челюсти недоразвиты:
о Ветвь и тело нижней челюсти сформированы пропорционально мыщелку
• Окклюзионная плоскость приподнята на пораженной стороне
• Иногда обнаруживается расщелина неба
• Скуловая кость и скуловая дуга уменьшены в размерах
• Гипоплазия сосцевидного отростка
• Дистопия глазницы: глазница смещена кнаружи и книзу
• Дефекты среднего и наружного уха
3. Рекомендации по визуализации:
• Лучший метод визуализации:
о КЛКТ, КТ
о Панорамные изображения для общей оценки челюстей
1. Перелом:
• Младенческий перелом мыщелка со смещением проксимального сегмента кпереди, книзу, кнутри и сращением в этом положении:
о Сужение и изменение формы сигмовидной вырезки
• Укорочение ветви и тела нижней челюсти
2. Гипоплазия мыщелка:
• Мыщелок нормальной формы, но уменьшен в размерах
• Уменьшение размеров ипсилатеральной половины нижней челюсти в зависимости от возраста дебюта и выраженности гипоплазии
3. Дегенеративное заболевание:
• Поражение одностороннее, дебютирует до полового созревания: мыщелок и ипсилатеральная половина нижней челюсти малого размера
(Слева) На фотографии (в профиль) пятилетней девочки с гемифациальной микросомией видна недоразвитая нижняя челюсть; правое ухо сформировано нормально. Нижняя челюсть отклонена в пораженную трону, из-за чего она недостаточно выстоит кпереди.
(Справа) На фотографии (анфас) у этой же пациентки определяется умень -шение размеров лица на стороне поражения, типичное для ГФМ. Обратите внимание на отклонение подбородка к пораженной стороне (влево) и низкое положение левого уха. (Слева) На панорамной реформатированной КЛКТ у этой же пациентки определяется, что нижняя челюсть и правый ВНЧС, включая мыщелок и суставную ямку, сформированы правильно.
(Справа) На панорамной реформатированной КЛКТ у этой же пациентки слева отсутствуют суставная ямка, суставное возвышение, а также мыщелок. Обратите внимание на очень короткую ветвь и скученные задние зубы нижней челюсти. Это ГФМ III типа. (Слева) На трехмерной реконструкции (КЛКТ, вид спереди) у этой же пациентки определяется дефект бокового отдела нижней челюсти. Низко расположен сосцевидный отросток.
(Справа) На корональной КЛКТ у этой же пациентки определяется низкое положение основания черепа слева. Смещение основания черепа книзу-типичное проявление при малом или отсутствующем мыщелке.
1. Общая характеристика:
• Этиология:
о Неизвестна; предполагается связь с гематомой в области 1-й и 2-й жаберной дуги или аномалией развития и миграции нервного гребня
2. Градации, классификация:
• Тип I: легкая гипоплазия мыщелка, ветви, тела
• Тип IIА: маленький плоский мыщелок, недоразвитие или отсутствие суставной ямки, маленькая ветвь неправильной формы, нормальное положение мыщелка в суставной ямке
• Тип IIВ: признаки, характерные для типа ПА, но с аномальным положением мыщелка (внизу, медиально, спереди)
• Тип III: несформированный ВНЧС, маленькая ветвь или ее отсутствие, маленькое тело нижней челюсти
1. Проявления:
• Типичные признаки/симптомы:
о Уши: микротия, преаурикулярные «метки» или ямки, атрезия слухового прохода о Лицо:
- Макростомия
- Асимметрия лица:
Отклонение подбородка в сторону поражения
Уменьшение размеров лица
- Челюсти и зубы:
Маленький мыщелок или его аплазия
Элевация окклюзионной плоскости
Импактные зубы на стороне поражения
2. Течение и прогноз:
• Нарушение или замедление роста на стороне поражения с развитием прогрессирующей или непрогрессирующей асимметрии
3. Лечение:
• Ортодонтическое и хирургическое
е) Диагностическая памятка:
1. Следует учесть:
• Подмыщелковый перелом со смещением, полученный при рождении
• Гипоплазию мыщелка
• Дебют дегенеративного заболевания сустава в предпубертатном периоде
2. Советы по интерпретации изображений:
• Асимметрия нижней челюсти (включая мыщелок, ветвь, тело) + поражение одной или обеих глазниц, скуловой кости, уха, жевательных мышц
ж) Список использованной литературы:
1. Choi JW et al: Three-dimensional functional unit analysis of hemifacial microsomia mandible-a preliminary report. Maxillofac Plast Reconstr Surg. 37(1):28, 2015
2. Modica RF et al: Goldenhar syndrome associated with extensive arterial malformations. Case Rep Pediatr. 2015:954628, 2015
Симптомы, лечение и причины гемифациальной микросомии
Заячья губа Операция гемифациальной Микросомии помогла многим пациентам вернуть себе счастливое лицо и улыбку. Операция по гемифациальной макросомии послужила возрождением для пациента, который страдал от лицевых деформаций и думал, что никогда больше не будет улыбаться и иметь свое обычное лицо.
Гемифациальная микросомия - это состояние, при котором одна сторона лица недоразвита и не растет нормально, а также поражает скулы, нижнюю челюсть, лицевые нервы и шею. Черепно-лицевая Микросомия-это аномалия, которая возникает у детей на лице и черепе еще до их рождения. Термин макросомия означает не что иное, как аномальную малость строения тела, которая у людей варьируется от размера к размеру, причем стороны могут быть либо левой стороной лица, либо правой стороной лица человека. Люди, страдающие от черепно-лицевой микросомии, имеют аномалии в ушах и имеют рост кожи перед ушами или закрытый или отсутствующий слуховой проход, а проблемы со зрением встречаются редко. Говорят, что люди, страдающие гемифациальной микросомией, имеют синдром Гольденхара.
Что такое Гемифациальная Микросомия?
Как мы знаем, гемифациальная микросомия-это врожденное заболевание, которое возникает случайно, но также наследуется в некоторых семьях. Гемифациальную микросомию путают с синдромом Гольденхара, который является редким врожденным заболеванием. Гемифациальная микросомия является одной из характеристик синдрома Гольденхара и включает аномалии позвоночника, эпибульбарные дермоиды и липодермоиды. Это асимметричная черепно-лицевая аномалия, поражающая строение от первой и второй дуг глотки. Гемифациальная микросомия характеризуется структурными аномалиями орбиты, верхней челюсти, нижней челюсти и черепных нервов. Гемифациальная микросомия была впервые использована в 1964 году Горлиным и Пиндборгом. Часто спрашивали, болезненна ли гемифациальная микросомия или нет.
Ответ на него-нет, и ребенок не жалуется ни на боль, ни на дискомфорт. Это расстройство, которое случается примерно с одним из 5000 новорожденных и может возникать спорадически. Некоторые проблемы возникают при гемифациальной микросомии, такой как паралич лица, истончение мягких тканей щек и недоразвитие глазницы. Лечение заболевания может быть направлено на устранение асимметрии челюстей, малого и отсутствующего уха, а также мягких тканей щек. Черепно-лицевая микросомия - это генетическое заболевание, которое вызвано мутацией и изменением определенного гена. Черепно-лицевая микросомия содержит симптомы, которые могут влиять на различные части черепа и лица. Ранняя диагностика и раннее лечение необходимы, если челюсть недоразвита и вызывает проблемы с дыханием и питанием. Таким же образом, в случае синдрома Гольденхара, который является тем же самым, что и гемифациальная микросомия, существует редкая врожденная инвалидность, характеризующаяся неполным развитием в таких частях, как уши, нос, мягкие пластины, губы и нижняя челюсть. Люди, имеющие проблемы с Гольденхаром столкнутся с такими проблемами, как проблемы с питанием, потеря слуха, трудности с дыханием, биполярные опухоли, черепно-лицевые аномалии и трудности в разговоре.
Другие названия для: Гемифациальный низкорослости
Другие названия гемифациальной микросомии называются такими, как краниофациальная микросомия, синдром Гольденхара, оромандибулярный дизостоз, Фацио-аурикула-вертебральный синдром, синдром первой и второй ветвящейся дуги, окуло-аурикула - вертебральный спектр и латеральная лицевая дисплазия, синдром первой и второй глоточной дуги, оромандибулярный дизостоз, HFM, синдром Гольденхара Горлина и фациоаурикуловертебральная дисплазия. Все это названия гемифациальной микросомии.
Какие Причины Гемифациального Низкорослости?
Для многих людей до сих пор причина гемифациальной микросомии неизвестна, и считается, что некоторые проблемы возникают в первые дни развития, такие как нарушение кровоснабжения первой и второй дуг ветвей на 7-8 неделе беременности. Существуют экологические факторы риска, такие как лекарства во время беременности, такие как аспирин или ибупрофен. Гемифациальная микросомия возникает как генетическое расстройство, такое как хромосомное расстройство, которое встречается в некоторых семьях. Можно сказать, что гемифациальная микросомия может возникать как сочетание генетических изменений и экологических рисков.

признаки и симптомы Гемифациальной Микросомии
Гемифациальная микросомия имеет следующие признаки и симптомы.
- Асимметрия лица
- Аномалии в наружном ухе
- Мелкие, уплощенные верхнечелюстные и скуловые кости
- Ушные бирки
- Аномалии развития зубов и задержка их развития
- Суженная челюсть
- Сокращение мышц лица
- Глухота в результате аномалий слуха
- Отсутствие мышц на щеках
- Дефекты скелета в том числе проблемы со спинным мозгом и ребрами
У детей с легкой формой гемифациальной микросомии челюсти меньше, а перед ухом имеется кожная метка. В тяжелых случаях лицо ребенка может показаться более скромным с одной стороны при ненормальной форме уха.
Как диагностируется Гемифациальная Микросомия?
Генетик, который имеет опыт, расстройство в специалисте, которое является результатом проблемы в генах, является правильным человеком для диагностики гемифациальной микросомии. Все эксперты не верят, что причина гемифациальной микросомии всегда связана с генетикой. История болезни ребенка будет изучена генетиком для диагностики гемифациальной микросомии. Существует несколько тестов для подтверждения диагноза, таких как компьютерная томография, рентген головы и компьютерная томография.
Как лечится Гемифациальная Микросомия?
Лечение гемифациальной микросомии варьируется в зависимости от имеющихся признаков и ее тяжести у каждого пострадавшего человека. Для лечения гемифациальной микросомии требуется много видов хирургических вмешательств. В некоторых случаях дети нуждаются в дыхательной поддержке вскоре после рождения, если челюсть сильно поражена, но хорошая новость заключается в том, что проблемы с дыхательными путями можно решить без хирургического вмешательства. Дети с деформациями челюстей могут испытывать трудности с кормлением и нуждаются в дополнительном кормлении, которое осуществляется через назогастральную трубку, поддерживающую рост и увеличение веса. Дети с отсутствующими ушами или аномально сформированными слуховыми аппаратами могут пройти множество реконструктивных операций, чтобы уши выглядели нормально. Аномалии костной ткани также могут потребовать хирургического вмешательства. При гемифациальной микросомии задействованы многие системы организма, поэтому за больными необходимо постоянно наблюдать. В случае, если у ребенка наблюдается паралич лица или аномалии век, закрытие глаза может быть неполным, и защита будет обеспечиваться либо с помощью смазочных материалов, либо хирургическим путем. Традиционное лечение гемифациальной микросомии включает ортогнатическую хирургию.
Содержание книги информировало нас о гемифациальном расстройстве микросомии, и это позволило нам узнать его типы и то, как мы можем лечить эту болезнь и дать детям лицо, которого они заслуживают, прямо в раннем возрасте. Хорошая жизнь со здоровым образом жизни-это то, что каждый заслуживает. Идите вперед и помогите кому-нибудь со знаниями, которые дал вам этот пост, чтобы вернуть улыбку на лицо человека, который был поражен этим расстройством.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
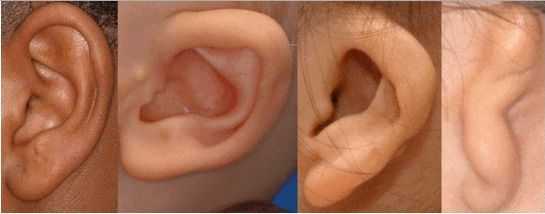
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
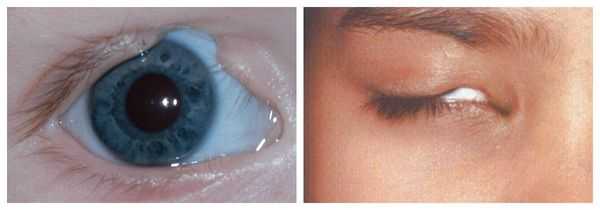
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
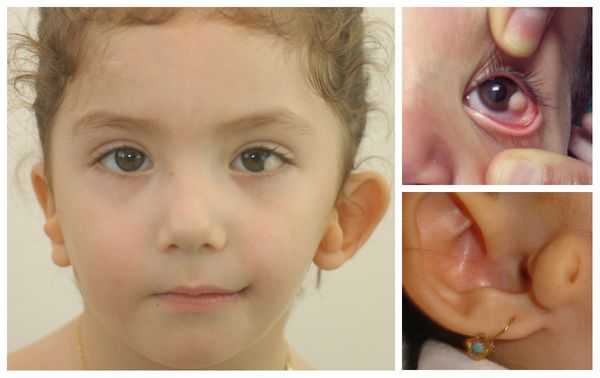
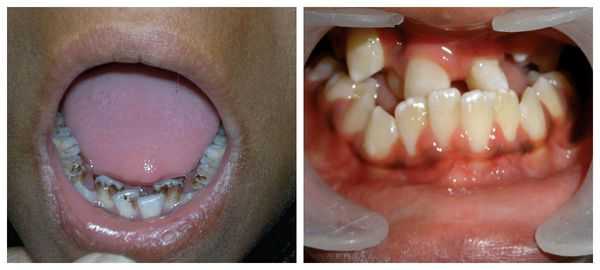
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
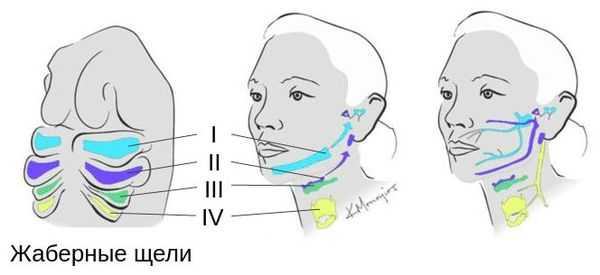
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
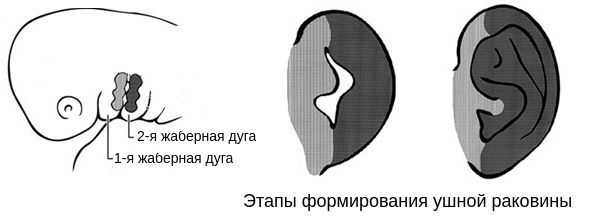
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
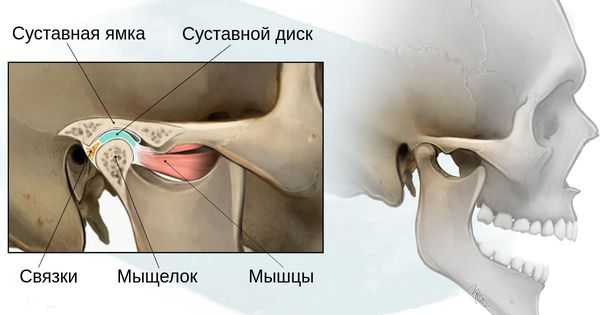
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
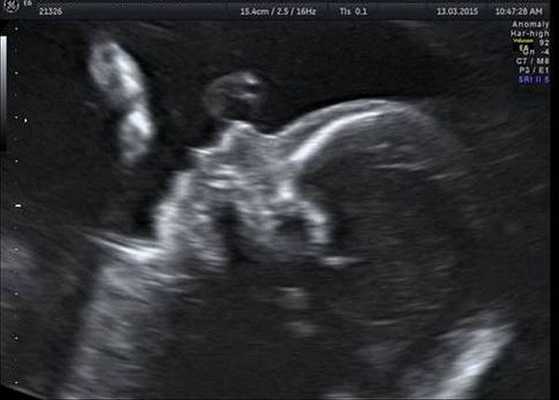
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
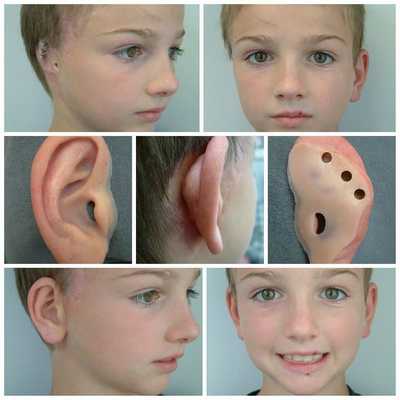
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
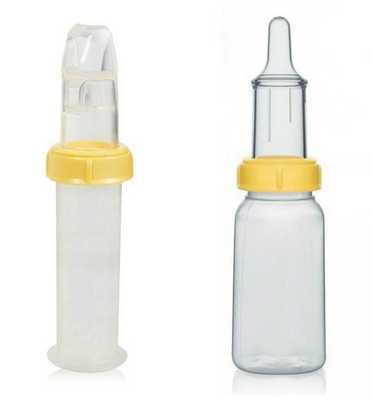
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Читайте также:
- Клинические варианты атеросклероза. Клинические проявления атеросклероза.
- Диагностика краниостеноза по КТ, МРТ
- Микропапулезный туберкулид. Милиарная диссеминированная волчанка лица
- Обзор аномалий рефракции
- Связи базальных ганглиев: двигательная петля, когнитивная петля, лимбическая петля, глазодвигательная петля