Ладонно-подошвенная кератодермия
Добавил пользователь Валентин П. Обновлено: 27.01.2026
В статье освещены сведения о кератодермиях – гетерогенной группе состояний, характеризующихся аномальным утолщением кожи ладоней и подошв. Традиционно выделяют приобретенные и наследственные формы.
Резюме. В статье освещены сведения о кератодермиях – гетерогенной группе состояний, характеризующихся аномальным утолщением кожи ладоней и подошв. Традиционно выделяют приобретенные и наследственные формы. В клинической практике наиболее часто встречается гиперкератоз ладоней и подошв как одно из проявлений псориаза, экземы, дерматомикозов и многих других заболеваний. К развитию гиперкератоза ладоней и подошв могут также привести механические и токсические факторы (в том числе прием лекарственных препаратов), поступление с пищей токсических веществ, приводящих к изменениям слизистой кишечника, современные требования моды и красоты могут способствовать развитию множественного дефицита витаминов. Значительно реже встречаются наследственные формы кератодермий, являющиеся самостоятельными заболеваниями. Раннее начало и семейный анамнез предполагают генетическую природу кератодермии. Отличительными особенностями наследственных форм служат характер наследования, степень поражения эпидермиса, наличие/отсутствие распространения очагов за пределы кожи ладоней и подошв, сопутствующая патология. В основе развития наследственных форм лежат мутации различных генов, кодирующих белки (например, кератин, десмосомы, лорикрин, катепсин С, белки щелевых контактов), которые принимают участие в процессе кератинизации. Наследственные ладонно-подошвенные кератодермии имеют большую генетическую и фенотипическую неоднородность, вследствие чего постановка точного диагноза на основе одних лишь клинических проявлений, когда нет возможности выполнить молекулярно-генетическое исследование, является весьма сложной задачей. Благодаря секвенированию нового поколения был достигнут значительный прогресс в расшифровке генетической основы кератодермий. В данном обзоре рассмотрены патогенетические, клинические, диагностические особенности диффузных форм кератодермий, варианты симптоматической терапии, учитывая торпидность и резистентность патологического процесса.
Кератодермии – гетерогенная группа заболеваний, характеризующаяся стойким утолщением эпидермиса кожи ладоней и подошв. После ихтиозиформных поражений кожи кератодермии занимают второе место в общей структуре генодерматозов. Заболевание чаще встречается у женщин, наследственная отягощенность колеб-лется от 1:1000 до 1:4000-9000 [1]. Выделяют наследственные и приобретенные ладонно-подошвенные кератодермии.
В клинической практике чаще всего встречаются приобретенные формы при псориазе, красном плоском лишае, питириазе, красном волосяном отрубевидном (болезни Девержи), экземе, ихтиозе, дерматомикозах, злокачественных новообразованиях и ряде других заболеваний. В ряде случаев присоединяется патологическая сухость кожи (чаще у женщин), нередко кератодермии имеют генетическое происхождение [2]. Также к развитию гиперкератоза ладоней и подошв могут привести механические и токсические факторы (в том числе прием лекарственных препаратов), поступление с пищей токсических веществ, приводящих к изменениям слизистой кишечника, современные требования моды и красоты могут способствовать развитию множественного дефицита витаминов, возможны и иные повреждающие факторы [3, 4]. Исключив вероятность развития приобретенной формы, необходимо задуматься о наследственной.
В настоящее время описано более 20 различных видов кератодермий, являющихся самостоятельными заболеваниями [3]. В основе развития наследственных форм лежат мутации различных генов, кодирующих белки (например, кератин, десмосомы, лорикрин, катепсин С, белки щелевых контактов), которые принимают участие в процессе кератинизации [5].
Варианты мутаций обуславливают клинический полиморфизм. Ряд кератодермий характеризуется лишь ограниченным поражением кожи ладоней и подошв, в то время как при других типах наблюдаются генерализованные нарушения кератинизации. Патологическим изменениям могут подвергаться придатки кожи (ногти и волосы), зубы, возможна потеря слуха, поражение глаз, развитие кардиомиопатии, злокачественных новообразований [6]. Вследствие хронического прогредиентного течения у пациентов формируется физический и психоэмоциональный дискомфорт.
Наследственные ладонно-подошвенные кератодермии имеют большую генетическую и фенотипическую неоднородность, вследствие чего постановка точного диагноза на основе одних лишь клинических проявлений, когда нет возможности выполнить молекулярно-генетическое исследование, является весьма сложной задачей для клиницистов [5].
Секвенирование нового поколения помогло обнаружить гены, неизвестные ранее. Выяснение механизмов развития заболевания открывает новые возможности для разработки альтернативных методов лечения, все больше вызывая интерес к данной патологии. Новые открытия стали предпосылкой к реклассификации некоторых ладонно-подошвенных кератодермий, которые учитывают не только клинические особенности, но и молекулярную основу.
Кератодермия Унны – Тоста (Unna – Thost palmoplantar keratoderma) наследуется по аутосомно-доминантному типу и является наиболее распространенной наследственной кератодермией с частотой развития 1:100 000 [6]. Описаны мутации в гене KRT9, расположенном на хромосоме 17q12-21, которые обуславливают тяжелый эпидермолиз. Ген KRT9 специфичен только для кожи ладоней и подошв, поэтому патологический процесс при данной кератодермии не распространяется за пределы ладонно-подошвенной области. Реже обнаруживается мутация в гене KRT1, при этом явления эпидермолиза минимальны либо отсутствуют совсем. Считается, что нарушение целостности промежуточных филаментов в результате этих мутаций уменьшает устойчивость цитоскелета к незначительной внешней травме, что приводит к образованию пузырей, гиперкератозу, а также к эпидермолизу.
В 2002 г. Кюстер и другие исследователи обнаружили эпидермолитический гиперкератоз по данным гистологии, что характерно для эпидермолитической кератодермии Вернера [7]. Генетическое тестирование выявило мутацию p.R162W в KRT9 в исходном семействе, отмеченную Унной и Тостом, и мутацию p.N160I в KRT9 в исходном семействе, отмеченную Вернером. Обе мутации были расположены на одном сегменте coli-1A в начале домена центрального стержня KRT9. Согласно данному открытию стало очевидно, что данные кератодермии являются одним и тем же типом [8]. Клинически такой тип кератодермии характеризуется избыточным ороговением кожи ладоней и подошв, при этом не наблюдается перехода на другие области. У детей на первом году жизни можно заметить легкое утолщения кожи, далее отмечается постепенное нарастание гиперкератоза. Как правило, к 4-5 годам патологический процесс приобретает вид грубых, толстых гиперкератотических наслоений желтого цвета, с резко ограниченным краем, окружен эритематозным венчиком шириной 1-3 мм. Процесс сопровождается глубокими трещинами, болезненностью, локальным гипергидрозом, нередко появлением пузырей [1].
Ряд авторов выявили рентгенологическую картину остеопороза и атрофии фаланг, наличие подвывихов и деформирующего артроза межфаланговых суставов кистей и стоп [5]. Ногти обычно подвергаются незначительным изменениям, могут иметь деформацию в форме песочных часов. Часто встречаются грибковые суперинфекции.
При гистологическом исследовании выявляется эпидермолитический гиперкератоз (акантокератолизис и дегенерация гранулярного слоя) [3].
Кератодермия Меледа (болезнь острова Меледа (Млета), акрокератома врожденная прогрессирующая, наследственная преходящая ладонно-подошвенная кератодермия, кератоз наследственный трансгредиентный и прогредиентный) является редкой формой кератодермий с предполагаемой распространенностью 1:100 000 [9]. Большинство случаев происходит в родственных кланах, это семьи из Хорватии, Алжира, Израиля и Туниса. Заболевание распространено по всему миру, особенно в регионах, которые исторически были торговыми путями Дубровницкой республики. Мутация распространилась в результате миграции и сохранилась только потому, что она не смертельна и не влияет на воспроизводство потомства [10]. Впервые о данном заболевании стало известно от дубровницкого врача Луки Стулли (Стулича), который описал его в итальянском журнале Antologia в 1826 г. Он наблюдал кератодермию у ряда своих островных пациентов, охарактеризовал ее как «ненатуральную структуру кожи». Незаразное наследственное кожное заболевание было названо mal de Meleda (млетская болезнь) в честь острова Меледа (Млет) в Адриатическом море. Остров сотни лет использовался как изолятор для больных чумой и лепрой, что способствовало созданию условий для кровного родства [11].
Болезнь острова Меледа характеризуется аутосомно-рецессивным типом наследования. Кератодермия обусловлена биаллельными мутациями в ARS-гене (расположен на хромосоме 8q24.3), кодирующем протеин SLURP1 (секретируемый белок Ly-6/PLAUR 1) [3]. Ген SLURP1 локализуется в зернистом слое эпидермиса и выполняет роль проаптотического белка. Арредондо и другие доказали, что белок SLURр1 связывается через никотиновые рецепторы ацетилхолина с кератиноцитами и приводит к уменьшению количества этих клеток. Соответственно, мутация гена, кодирующего данный белок при кератодермии Меледа, приводит к нарушению его функции, регуляция апоптоза кератиноцитов меняется, и возникает гиперкератоз. Наличие хронического воспалительного инфильтрата можно объяснить еще одной функцией белка SLURP1. Считается, что он ингибирует высвобождение макрофагами и кератиноцитами фактора некроза опухоли альфа (ФНО-α). Из-за отсутствия функционального белка SLURP1 процесс не регулируется должным образом. Высвобождение ФНО-α в эпидермисе в конечном итоге приводит к хемотаксису дендритных клеток и клеток памяти, поддерживающих воспаление [11]. Клинические признаки, как правило, включают двустороннюю ладонно-подошвенную кератодермию, развитие диффузного гиперкератоза в форме перчаток или носков с резкими границами и желтым оттенком. Начало болезни характеризуется появлением стойкой эритемы с шелушением кожи ладоней и подошв, как правило, в раннем детском возрасте. Далее, обычно к 15-20 годам, наблюдается усиление ороговения. Характерны участки желто-коричневого гиперкератоза, которые с возрастом переходят на тыльную поверхность кистей и стоп, область локтевых и коленных суставов. Края очага поражения очерчены каймой с фиолетовым оттенком, ширина которой составляет несколько миллиметров. Выражен локальный гипергид-роз, в результате чего поверхность пораженных участков становится слегка влажной, с черными точками (вывод-ные протоки потовых желез). В периоральной области наблюдаются стойкая шелушащаяся эритема, небольшая инфильтрация. Кератодермия часто сопровождается дистрофией ногтей (койлонихия, подногтевой гиперкератоз, поперечная или продольная исчерченность, вдавления, онихогрифоз), характерны сочетания болезни острова Меледа с атопией, возможно присоединение бактериальной и грибковой инфекции. В некоторых случаях кератоз сочетается с умственным или физическим недоразвитием, у отдельных больных наблюдались изменения на электроэнцефалограмме [9]. Гистологически в эпидермисе выявляют гиперкератоз, иногда акантоз без признаков эпидермолиза. Может наблюдаться паракератоз, были описаны случаи с характерной картиной гипергранулеза. В сосочковом слое дермы – небольшой хронический воспалительный инфильтрат из лимфоцитов и гистиоцитов [11].
Синдром Папийона – Лефевра (ладонно-подошвенный гиперкератоз с периодонтитом) – редкое заболевание, впервые описанное в 1924 г. Считается, что синдром поражает от одного до четырех человек на миллион [12]. Наследуется по аутосомно-рецессивному типу. Синдром Папийона – Лефевра обусловлен мутациями гена CTSC (расположен на хромосоме 11q14.2), кодирующего катепсин C (также известный как дипептидилпептидаза I). CTSC представляет собой олигомерную лизосомальную цистеиновую протеазу, которая играет важную роль в эпидермальной дифференцировке, а также активации серин-протеаз, вырабатываемых клетками иммунной системы. Мутации гена CTSC приводят к практически полной потере активности катепсина C, которая, по всей видимости, является причиной восприимчивости к определенным вирулентным возбудителям. Сообщалось о других клеточных нарушениях (включая окислительный стресс с нарушением антиоксидантной активности), характеризующихся аномально высокими уровнями гидропероксида и измененным содержанием CoQ и витамина E. Кроме того, снижается антимикробная активность нейтрофилов. Периодонтит обусловлен совокупностью факторов, таких как свободные радикалы, активные формы кислорода и митохондриальная дисфункция [9]. Развивается в возрасте от 1 до 4 лет, при этом проявления, как правило, сильнее на коже подошв, чем на ладонях. Начинается с эритемы, далее развивается гиперкератоз, который носит диффузный характер. В дальнейшем очаги гиперкератоза переходят трансгредиентно на тыл кистей и стоп. На локтях и коленях появляются очаги, напоминающие псориазиформные элементы. Наряду с поражением кожи развивается выраженный гингивит, он с большой скоростью прогрессирует, переходит в пародонтит, который сопровождается лизисом костей альвеолярных отростков, приводит к преждевременной потере молочных зубов. На фоне переохлаждения, во время развития острого пародонтита наступает обострение кожного процесса. Отмечено несколько случаев синдрома Папийона – Лефевра с неострым пародонтитом и/или пародонтитом с поздним началом. То же самое происходит и с постоянными зубами, обычно к 14-15 годам пациенты с данным синдромом все беззубые [13].
Большая часть пациентов с синдромом Папийона – Лефевра имеет повышенную восприимчивости к генерализованным инфекциям и инфекциям кожи (пиодермии). Описаны случаи развития абсцесса печени. Первые данные об этом были опубликованы в 1988 г. Патогенные бактерии обычно достигают печени гематогенным путем. Наиболее частым этиологическим агентом является золотистый стафилококк. Следует учитывать риск развития гнойного абсцесса печени у пациентов с синдромом Папийона – Лефевра, ассоциированного с лихорадкой неясного генеза [14]. У пациентов может развиваться гипергидроз со зловонным запахом, фолликулярный гиперкератоз, дистрофия ногтей, обызвествление твердой мозговой оболочки. В редких случаях прослеживается связь синдрома Папийона – Лефевра со злокачественной меланомой или плоскоклеточным раком. Гистологически выявляют утолщение всех слоев эпидермиса, особенно рогового, в дерме – незначительные клеточные скопления лимфоцитов и гистиоцитов.
Дифференциальная диагностика диффузных форм наследственных кератодермий проводится со схожими по клинической картине заболеваниями. Для каждой формы кератодермии характерны свои отличительные особенности. При проведении дифференциальной диагностики учитывают характер наследования, генетический дефект, степень поражения эпидермиса, наличие/отсутствие распространения очагов за пределы кожи ладоней и подошв, сопутствующую патологию. Сравнительная характеристика представлена в табл.
Диагностика кератодермий включает сбор анамнеза (с особым вниманием к семейному), физикальный осмотр, включающий оценку поражения кожных покровов, ногтей, волос и зубов, а также клиническую картину. Диагноз устанавливают при наличии очагового или диффузного гиперкератоза на ладонях и подошвах. Дебют заболевания в детском возрасте, отягощенный семейный анамнез, стойкая клиническая картина с небольшим изменением симптомов, относительная резистентность к терапии – это признаки, как правило, свидетельствующие о наследственной форме кератодермии. Отрицательный семейный анамнез или начальные проявления во взрослом возрасте не исключают ее возможности. При необходимости проводится гистологическое исследование.
Помимо неспецифических проявлений, таких как гиперкератоз, гранулез и акантоз, можно выявить характерные признаки, такие как эпидермолитический гиперкератоз, нарушение адгезии кератиноцитов (свидетельствует о десмосомных дефектах) или паракератоз (характерный для лорикриновой кератодермии). Разграничение эпидермолитических и неэпидермолитических форм ладонно-подошвенных кератодермий имеет терапевтическое значение, учитывая, что эпидермолитические формы на фоне приема системных ретиноидов имеют тенденцию к ухудшению [15]. Генетическое тестирование позволяет установить точный диагноз. В случае пародонтоза при кератодермии Папийона – Лефевра пациент направляется на консультацию к стоматологу, где стоматологическая рентгенография позволяет выявить атрофию альвеолярной кости.
Современные методы лечения наследственных форм кератодермий представляют собой в основном симптоматическую терапию. Основными принципами терапии являются увлажнение, восстановление гидролипидной мантии и отшелушивание роговых наслоений. Даются общие рекомендации пациентам: регулярные ванны, очищение и увлажнение области ороговения; избегать длительной механической нагрузки на кожу ладоней и подошв; носить ортопедическую обувь, использовать стельки. В качестве местной терапии применяются кератолитические средства (мочевина, салициловая кислота, молочная кислота). Возможно применение данных лекарственных препаратов под окклюзионную повязку на ночь [16]. При системной терапии используют препараты ретинола (витамина А). Чаще всего это ацитритин (0,3-1,0 мг/кг в зависимости от тяжести процесса). Ретиноиды регулируют процессы роста и трансформации клеток, оказывают терапевтический эффект путем модуляции дифференцировки кератиноцитов, подавления гиперпролиферации и уменьшения инфильтрации воспалительными клетками. Для этого используются синтетические ретиноиды, фитоэстрогены. Клинический эффект наблюдается не сразу, а по истечении 5-6 месяцев после начала приема препаратов и прекращается после их отмены [17]. Ретиноиды обычно применяют для лечения тяжелых, приводящих к инвалидности ладонно-подошвенных кератодермий. Ограниченность применения обусловлена большим количеством побочных эффектов (тератогенность, сухость слизистых оболочек, головная боль, повышение уровня в сыворотке крови холестерина, триглицеридов, трансаминаз и др.). У пациентов с эпидермолитической кератодермией лечение ретиноидами может привести к усилению образования пузырей [16]. Дополнительно используется профессиональная гигиена полости рта, проводится системная антибактериальная терапия периодонтита у пациентов с кератодермией Папийона – Лефевра. При присоединении бактериальной и грибковой флоры необходима наружная терапия антибактериальными и противогрибковыми средствами.
Таким образом, детальное изучение механизмов развития патогенетических, клинических, диагностических особенностей кератодермий открывает новые возможности для разработки методов лечения, повышая интерес к данной патологии.
КОНФЛИКТ ИНТЕРЕСОВ. Авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить.
CONFLICT OF INTERESTS. Not declared.
Литература/References
КГМА – филиал ФГБОУ ДПО РМАНПО Минздрава России; 420012, Россия, Казань, ул. Толстого, 4, корп. 2
Сведения об авторах:
Information about the authors:
Ладонно-подошвенная кератодермия
Московский научно-практический Центр дерматовенерологии и косметологии Департамента здравоохранения, Москва
Московская медицинская академия им. И.М. Сеченова
Московский научно-практический Центр дерматовенерологии и косметологии Департамента здравоохранения Москвы, 119071, Москва, Российская Федерация
Патогенетические аспекты, лежащие в основе ладонно-подошвенных кератодермий. Современные методы терапии
Журнал: Клиническая дерматология и венерология. 2015;14(2): 17‑23
Московский научно-практический Центр дерматовенерологии и косметологии Департамента здравоохранения, Москва
Ладонно-подошвенные кератодермии представляют собой гетерогенную группу болезней наследственного или приобретенного характера, в клинической картине которых присутствуют диффузные или очаговые утолщения рогового слоя эпидермиса преимущественно в области ладоней и подошв, реже в сочетании с кератозами другой локализации. Для них характерны эктодермальная дисплазия и воспаление. В статье представлены общие сведения об эпидемиологии, этиологии и патогенезе данной патологии; дана классификация наследственных кератодермий, представлена связь генетических мутаций с проявлениями основных синдромов. Описаны клинические проявления кератодермий как приобретенного, так и наследственного характера. Авторы обращают внимание на то, что терапевтические мероприятия при кератодермиях чаще носят симптоматический характер. Для общего лечения всех форм кератодермий показано применение ретиноидов, предпринимаются попытки применения заместительной гормональной терапии. Наружное лечение кератодермий включает бальнеолечение, использование кератолитических и увлажняющих топических средств. В комплексе с наружной терапией с успехом применяются физиотерапевтические методы. Учитывая, что кератодермии в ряде случаев могут выступать симптомами паранеопластических процессов и других заболеваний, необходимо продолжить диагностический поиск и дифференцированный подход к терапевтическим мероприятиям.
Московский научно-практический Центр дерматовенерологии и косметологии Департамента здравоохранения, Москва
Московская медицинская академия им. И.М. Сеченова
Московский научно-практический Центр дерматовенерологии и косметологии Департамента здравоохранения Москвы, 119071, Москва, Российская Федерация
Ладонно-подошвенные кератодермии (ЛПК) (кератоз ладонно-подошвенный) составляют достаточно гетерогенную группу болезней наследственного или приобретенного характера, в клинической картине которых присутствуют диффузные или очаговые утолщения рогового слоя эпидермиса преимущественно в области ладоней и подошв, реже в сочетании с кератозами другой локализации. Характерна эктодермальная дисплазия и воспаление [1—3].
Данная патология, являясь одной из наиболее распространенных среди дискератозов, в то же время занимает II место в структуре наследственных заболеваний кожи. Распространенность ладонно-подошвенных кератодермий зависит от множества факторов: климатогеографических зон, этнического разнообразия популяций и вариабельности условий окружающей среды [4]. В европейских странах среднестатистическая распространенность кератодермий составляет 1:2500. В странах Африки и Латинской Америки она значительно ниже. При эпидемиологическом и медико-генетическом изучении, которое было проведено в различных регионах мира, в том числе и в России, наследственная отягощенность составила от 1:1000 до 1:4000—9000 [5].
Кератодермии чаще встречаются у женщин, при этом со временем года не связаны, в большинстве случаев развиваются в течение первого года жизни, но могут существовать с самого рождения или возникнуть во взрослом состоянии [4, 5].
По происхождению различают приобретенные и наследственные кератодермии. Наследственные кератодермии обусловлены мутациями в генах, кодирующих образование кератинов и белков клеточной оболочки. Они могут быть самостоятельными заболеваниями или сочетаться с разнообразными аномалиями, чаще всего эктодермального происхождения [1, 6]. Различают кератодермии с аутосомно-доминантным (А-Д) (кератодермия Тоста—Унны, кератодермия эпидермолитическая, кератодермия мутилирующая Фовинкеля, кератодермия Бушке—Фишера, кератодермия Сименса) или аутосомно-рецессивным (А-Р) (болезнь острова Меледа, кератодермия Папийона—Лефевра) типом наследования [7].
В практике врача дерматовенеролога довольно часто встречаются пациенты с приобретенными формами ЛПК, возникающими на фоне основного заболевания или вследствие внешних триггеров. Они сопровождают такие дерматозы, как красный волосяной лишай Девержи, красный плоский лишай, экзема, псориаз, ихтиоз. Кератодермии встречаются и при инфекционных заболеваниях — вторичном сифилисе, гонорейном кератозе, при синдроме Рейтера, дерматомикозах, норвежской чесотке, а также при нарушениях функции нервной и эндокринной систем, метаболических нарушениях (беременность, менопауза, гипотиреоз, микседема) [8]. Кератодермии могут быть маркерами паранеопластических процессов во внутренних органах. Описаны случаи, когда кератодермия сочеталась с такими онкологическими заболеваниями, как рак пищевода или желудка — развивался параонкологический кератоз ладоней и подошв. Возможно развитие профессиональных кератодермий, возникающих под воздействием лучевых, химических, механических факторов [9, 10].
По клиническим признакам выделяют две основные группы кератодермий — диффузные, при которых наблюдается сплошное поражение поверхностей ладоней и подошв, и очаговые, при которых участки избыточного ороговения располагаются островками, линейно, точечно, но не распространяются на всю поверхность (табл. 1) [11].

Таблица 1. Клиническая классификация наследственных кератодермий
Кератодермия может являться одним из основных симптомов ряда синдромов [12—17] (табл. 2).
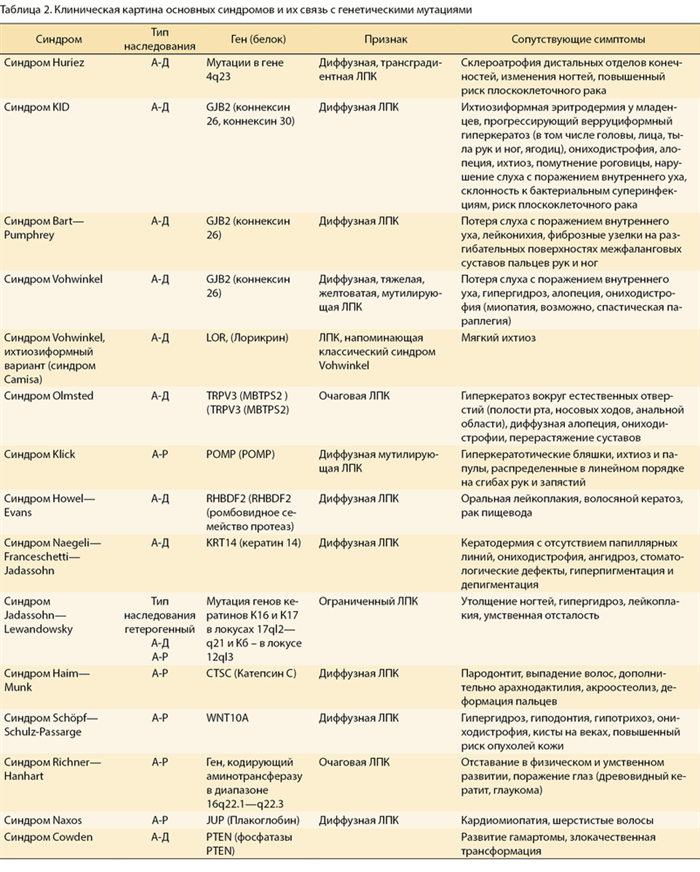
Таблица 2. Клиническая картина основных синдромов и их связь с генетическими мутациями
Все ЛПК, несмотря на многообразие клинических форм, имеют общие гистологические признаки: значительный гиперкератоз, реже акантоз и мононуклеарная инфильтрация верхнего слоя дермы, преимущественно вокруг кровеносных сосудов [18].
Как наследственные, так и приобретенные кератодермии характеризуются хроническим прогредиентным течением, торпидным к терапии. У таких пациентов имеются определенные затруднения при выборе профессии, устройстве на работу, при выполнении бытовых и профессиональных обязанностей. Больные кератодермиями, подавляющее большинство из которых женщины, испытывают выраженные психоэмоциональные расстройства, проблемы в личной жизни, у них значительно снижается самооценка. Все это приводит к ухудшению качества жизни и снижению социального статуса, оказывает негативное влияние на материальное благосостояние, а в особо тяжелых случаях следствием заболевания может стать инвалидность [19].
Наиболее часто в практике врача дерматовенеролога встречается наследственная диффузная кератодермия Унны—Тоста (тип А-Д), которая характеризуется возникновением массивных роговых наслоений на ладонях и подошвах. Дерматоз манифестирует в первые годы жизни в виде легкого утолщения кожи ладоней и подошв. Постепенно диффузный кератоз нарастает. Роговые наслоения гладкие, желтого цвета, с резко очерченным краем и эритематозным венчиком по периферии. Одновременно наблюдается локальный гипергидроз, гиперонихии, редко могут отмечаться множественные липомы, дистрофия роговицы и олигофрения. Возможно развитие остеопороза и остеолиза фаланг, деформирующего артроза межфаланговых суставов [20].
Кератодермия Меледа (кератоз наследственный трансградиентный) — редкая форма наследственной диффузной кератодермии (тип наследования А-Р), которая возникает в раннем детском возрасте, часто у детей от близкородственных браков. Характерным является переход кератоза на тыл кистей, стоп, области локтевых, коленных суставов (трансградиентный кератоз), локальный гипергидроз. Поверхность очагов кератоза при этом обычно влажная, что вызывает мацерацию и размягчение роговых наслоений. Участки пораженной кожи испещрены многочисленными трещинами, иногда кровоточащими и болезненными, проникающими до мальпигиевого слоя и даже собственно кожи. Характерны сочетание процесса с атопическим дерматитом, осложнения пиококковой инфекцией, дистрофия ногтей с их резким утолщением или койлонихией. Могут отмечаться умственная отсталость, синдактилия, складчатый язык, готическое небо [19].
Кератодермия Папийона—Лефевра — наследственная диффузная кератодермия (тип наследования А-Р), сочетающаяся с пиогенными инфекциями кожи и десен, пародонтозом, дистрофией альвеолярных отростков челюстей и, как следствие, почти полным выпадением зубов и аномалией их развития. Клиническая картина развивается в первые 5 лет жизни в виде эритемы, роговых наслоений, выраженность которых постепенно усиливается. Участки кератоза нередко выходят на тыл кистей и стоп, область пяточного (ахиллова) сухожилия, коленных и локтевых суставов. Характерен локализованный гипергидроз. Ногти дистрофичны, волосы не изменены [21—23].
Кератодермия эпидермолитическая — достаточно редкая форма наследственной диффузной кератодермии с аутосомно-доминантным типом наследования. Характеризуется эпидермолитическим гиперкератозом. Клинически данная форма очень сходна с кератодермией Унны—Тоста и отличается только гистоморфологическими признаками. Дериваты кожи не изменены, локальный гипергидроз отсутствует.
Кератодермия мутилирующая Фовинкеля (синонимы: наследственная мутилирующая кератома, синдром Фонвинкеля) — диффузная кератодермия (тип наследования А-Д), которая развивается на 1—2-м году жизни в виде роговых наслоений с гипергидрозом. В дальнейшем формируются «шнуровидные борозды» на пальцах, приводящие к контрактурам и их спонтанной ампутации. На тыльной поверхности кистей, а также в области локтевых и коленных суставов выражен фолликулярный кератоз, возможны кератотические бляшки на коже спины и ягодиц. Редко наблюдаются рубцовая алопеция, потеря слуха, изменение ногтей (часто по типу часовых стекол) [24—26].
Кератодермия диссеминированная Бушке—Фишера—Брауэра (син: симметричная эритематозная кератодермия Бенье, кератоз точечный рассеянный Бушке—Фишера) — самая распространенная форма очаговой наследственной кератодермии (тип наследования А-Д). Первые симптомы дерматоза появляются в возрасте от 15 до 30 лет. Клиническая картина характеризуется множественными участками ороговения желто-коричневого цвета, расположенными симметрично на коже ладоней, подошв и сгибательной поверхности пальцев. Очаги поражения не сливаются между собой, потоотделение не нарушено [27, 28].
Кератодермия Сименса относится к достаточно редким формам очаговой кератодермии, которая развивается в первые 10 лет жизни и проявляется очагами эритемы, которые формируются исключительно на местах наибольшего давления на коже подошв (чаще в области пятки), ладоней. Впоследствии в этих зонах развиваются округлые или неправильных очертаний участки кератоза размером от 1 до 5 см, затрудняющие ходьбу и сопровождающиеся выраженной болезненностью [29].
Кератодермия линеарная Фукса — форма кератодермии, отличающаяся линеарными участками кератоза ладоней и подошв. Тип наследования — аутосомно-доминантный. Заболевание начинается в течение первых нескольких лет жизни. Характеризуется наличием островков роговых наслоений вытянутой (линеарной) формы, расположенных на коже ладоней и подошв, а также в виде роговых гребней вдоль сухожильных влагалищ, выступающих на 5—10 мм над уровнем окружающей кожи. В процесс могут быть вовлечены ногти и волосы. Типичен гипергидроз и арковидное небо [19].
В ходе изучения литературы по данному вопросу выяснилось, что в практике врача-дерматовенеролога довольно часто имеет место обращение пациентов с приобретенными формами ЛПК, критериям которых соответствуют такие формы, как климактерическая кератодермия Хакстхаузена, кератодермия краевая ладоней Рамос-и-Сильвы, инфекционная кератодермия.
Кератодермия климактерическая (болезнь Хакстхаузена) — одна из самых распространенных форм приобретенной диффузной кератодермии, развивается у 10—15% женщин в климактерическом периоде, чаще в местах давления и трения, обычно сочетается с метаболическим синдромом, гипертонической болезнью, деформирующим артрозом [30]. Основным триггерным фактором в развитии этой кератодермии считается дефицит эстрогенов, прогестерона, тестостерона, развивающийся в процессе возрастной инволюции репродуктивной системы. У пациенток отмечают повышение продукции фолликулостимулирующего и лютеинизирующего гормонов на фоне снижения эстрадиола. Возможно опосредованное влияние снижения функции щитовидной железы, активируемой эстрогенами. Вместе с тем далеко не у всех женщин, вступивших в период климакса, даже тяжело протекающего, развивается климактерическая кератодермия. По-видимому, для развития заболевания необходимы дополнительные условия. Возможно, это уменьшение количества или функциональной активности рецепторов к эстрогенам в клетках кожи. Заболевание начинается с появления трещин, затем участков гиперкератоза. При прогрессировании болезни папулы сливаются в бляшки крупных размеров желтовато-коричневого цвета с нечеткими границами, которые покрываются толстыми гиперкератотическими наслоениями. Процесс носит симметричный характер. Жжение, зуд и боли при физической нагрузке связаны с наличием трещин. Заболевание имеет прогрессирующее течение, протекает волнообразно с периодами обострения, чаще в зимнее время [30]. Достижение полной ремиссии наблюдается редко. Облегчение состояния в ряде случаев наступает после окончания климактерического периода, на фоне длительной эстрогенотерапии или в результате лечения ретиноидами. В установлении диагноза климактерической кератодермии основными критериями являются характерная клиническая картина, данные гистологического исследования кожи, гинекологический анамнез, анализ течения заболевания и результаты предшествующей терапии. Данную патологию необходимо тщательно дифференцировать от микоза ладоней и стоп, хотя возможно и сочетанное поражение.
Инфекционная кератодермия возникает во время или после перенесенного инфекционного заболевания и носит преходящий характер, не требуя активного лечения.
Кератодермия краевая ладоней Рамос-и-Сильвы локализуется по краям ладонной поверхности кистей и чаще всего является признаком злокачественных новообразований [31].
Диагностика как наследственных, так и приобретенных кератодермий в связи с их гетерогенностью и редкостью отдельных форм часто представляет значительные трудности, и период от появления первых симптомов до установления точного диагноза, назначения адекватной терапии может быть долгим и обременительным для пациента. Диагноз «ладонно-подошвенная кератодермия» без уточнения ее нозологической принадлежности является неполным. При диагностике ЛПК учитывают характер процесса (ограниченный или диффузный), вовлечение других участков кожи, помимо ладоней и подошв (трансградиентные формы ЛПК), наследственность, возраст манифестации процесса, наличие симптомов со стороны других органов (например, ЛПК как часть какого-либо синдрома). Диагноз основывается на клинических признаках, лабораторных исследованиях, а при необходимости проводится гистологическое исследование.
Дифференциальный поиск включает различные формы кератодермий, псориаз, болезнь Девержи, дисгидротическую и тилотическую экзему, микозы.
Развитие приобретенной кератодермии, как правило, наблюдается в более позднем возрасте у пациентов без отягощенного семейного анамнеза. В некоторых случаях улучшение течения ЛПК во время отдыха указывает на профессиональный характер дерматоза. Другими данными являются наличие аллергии или инфекции, а также воздействие некоторых химических веществ (например, мышьяка, хлорированного углеводородного раствора для инъекций) или побочные эффекты от некоторых лекарств (например, бета-глюкана, лития, химиотерапевтических агентов) [32].
Подозрение на наследственную ЛПК должны вызывать следующие признаки: начало заболевания в детском возрасте, наличие отягощенного семейного анамнеза, торпидное течение без положительной динамики. Отрицательный семейный анамнез или начальные проявления во взрослом возрасте не исключают возможность наследственной ЛПК. При подозрении на наследственную природу заболевания проводятся молекулярно-генетические методы для идентификации генов, содержащих мутации, позволяющие провести точную генетическую классификацию [33].
На сегодняшний день, несмотря на достижения медицинской науки и успехи в изучении этиологии и патогенезе кератодермий, их лечение является достаточно сложным и длительным процессом. К сожалению, даже применение комплексного лечения с использованием самых современных медикаментозных препаратов не позволяет добиться полного излечения данного заболевания и окончательного выздоровления.
Терапевтические мероприятия при кератодермиях чаще носят симптоматический характер. Для лечения всех форм кератодермий показано назначение ретиноидов, которые регулируют процессы пролиферации и дифференциации клеток, оказывают терапевтический эффект путем модуляции дифференцировки кератиноцитов, подавления гиперпролиферации и уменьшения инфильтрации воспалительными клетками. Используется ацитретин в дозе не более 1 мг на 1 кг массы тела в сутки в течение до 2 мес. Возможно назначение длительных курсов витамина, А в дозировке по 100–150 тыс. МЕ в сутки в течение 1,5—2 мес.
Для лечения климактерической кератодермии в последнее время прибегают к заместительной гормональной терапии (фитоэстрогены). Клинический эффект наблюдается не сразу, а по истечении 5—6 мес после начала приема препарата и прекращается после его отмены. Однако эти препараты вызывают ряд негативных эффектов и имеют достаточно много противопоказаний. Поэтому применение фитогормонотерапии в лечении климактерической кератодермии на сегодняшний день дискутируется. При кератодермии Папийона—Лефевра показана обязательная санация полости рта и прием антибиотиков.
Наружное лечение кератодермий включает бальнеолечение, использование кератолитических и увлажняющих топических средств. Для наружной терапии также используют мази, содержащие витамины А, Е, регенерирующие препараты, а в ряде случаев — мази с глюкокортикоидами. Лечение назначают с учетом формы кератодермии, индивидуальных особенностей. Важна профилактическая направленность, в частности применение топических антибактериальных и антимикотических средств [1].
В комплексе с наружной терапией с успехом применяют физиотерапевтическое лечение. Положительный эффект отмечается при использовании ультрафонофореза витаминов, А и Е, криотерапии [34, 35]. Имеются данные об эффективности гелий-неонового лазера, углекислотного лазера, воздействие которыми проводится после предварительного удаления роговых масс [36, 37].
Таким образом, на сегодняшний день ЛПК представлены достаточно гетерогенной группой дерматозов — наследственных или приобретенных, которые в ряде случаев могут выступать симптомами паранеопластических процессов и других заболеваний, что предопределяет необходимость дальнейшего диагностического поиска и дифференцированного подхода к терапевтическим мероприятиям.
Кератодермия
Кератодермии - группа дерматозов, характеризующихся нарушением процессов ороговения, - избыточным рогообразованием преимущественно ладоней и подошв.
Причины и патогенез заболевания окончательно не выяснены. Исследованиями установлено, что кератодермии обусловлены мутациями в генах кодирующих кератин 6, 9, 16. В патогенезе большое значение имеют недостаточность витамина А гормональные дисфункции, в первую очередь половых желез, бактериальные и вирусные инфекции. Они являются одним из симптомов наследственных болезней и опухолей внутренних органов (перапсориатические кератодермии)
По характеру клинической картины кератодермии могут быть диффузными и локализованными. Кератодермия может быть одним из симптомов ряда синдромов и заболеваний.
Если Вас беспокоят вышеописанные симптомы, пройдите обследование в Клинике №1
Консультация дерматолога + анализы и процедуры (при наличии направления врача) со скидкой 20%.
Кератодермия Унны-Тоста - распространенная форма наследственной диффузной кератодермии, для которой характерен кератоз ладоней и подошв без перехода на другие участки кожи, окруженный эритематозным венчиком шириной 1-3 мм, локальный гипергидроз. Тип наследования аутосомно-доминантный.
Кератодермия Меледа - форма наследственной диффузной кератодермии, отличающаяся переходом кератоза с ладонно-подошвенных поверхностей на тыл кистей, стоп, области локтевых, коленных суставов (трансградиентный кератоз). Описана впервые среди кровных родственников населения острова Меледа. Тип наследования обычно аутосомно-рецессивный.
Кератодермия Папийона-Лефевра - наследственная диффузная кератодермия, сочетающаяся с парадонтозом и пиогенными инфекциями кожи и десен. Тип наследования аутосомно-рецессивный.
Кератодермия эпидермолитическая - редкая форма наследственной диффузной кератодермии. Отличается наличием признаков эпидермолитического гиперкератоза. Тип наследования аутосомно-доминантный.
Кератодермия мутилирующая Фовинкеля - форма наследственной диффузной кератодермии, сопровождающейся образованием фиброзных перетяжек, приводящих к ампутации пальцев. Тип наследования аутосомно-доминантный.
Кератодермия диссеминированная Бушке-Фшиера-Брауэра - наиболее распространенная форма очаговой наследственной кератодермии. Тип наследования аутосомно-доминантный. Первые симптомы болезни появляются в пубертатном периоде или несколько позже (от 15 до 30 лет). На коже ладоней, подошв и сгибательной поверхности пальцев появляются роговые узелки - "жемчужины", которые превращаются в плотные роговые желтовато-коричневые пробки с кратерообразным краем.
Кератодермия линеарная Фукса - форма наследственной очаговой кератодермии, отличающаяся линеарными участками кератоза ладоней и подошв. Тип наследования аутосомно-доминантный.
Кератодермия краевая ладоней Рамос-и-Сильвы - форма приобретенной ограниченной кератодермии, проявляющейся роговыми наслоениями по краю ладонных поверхностей. Развивается у больных со злокачественными новообразованиями внутренних органов, артритами, а также при нарушениях функции половых желез.
Кератодермия климактерическая (Хакстхаусена синдром) - форма приобретенной диффузной кератодермии, развивающаяся у женщин в климактерическом периоде. Первые симптомы болезни появляются на 5-м десятилетии жизни в виде эритемы на коже подошв, покрывающейся роговыми наслоениями, количество которых постепенно нарастает. Затем роговые наслоения появляются в центральных зонах ладоней. Состояние особенно ухудшается в зимнее время.
Если Вас беспокоят вышеописанные симптомы, пройдите обследование в Клинике №1
Консультация дерматолога + анализы и процедуры (при наличии направления врача) со скидкой 20%.
Лечение Кератодермии
В общей терапии кератодермии показан неотигазон. Доза препарата зависит от тяжести процесса и составляет 0,3-1 мг/кг веса больного. При отсутствии неотигазона рекомендуют витамин А в дозе от 100 до 300 000 мг в сутки длительное время. Наружная терапия заключается в использовании мазей с ароматическими ретиноидами, кератолитических и стероидных средств.
Клинический случай ладонно-подошвенной кератодермии Унны-Тоста Текст научной статьи по специальности «Клиническая медицина»
Аннотация научной статьи по клинической медицине, автор научной работы — Пашинян Альбина Гургеновна, Ильенко Л.И., Акопян А.Н.
Ладонно-подошвенные кератодермии гетерогенная группа наследственных дерматозов , которые могут быть самостоятельными заболеваниями или сочетаться с различными врожденными пороками развития, чаще всего эктодермального происхождения. Рассматривается дифференциальная диагностика, отмечаются характерные клинические признаки для каждой формы ладонно-подошвенной кератодермии . Кератодермию Унны-Тоста относят к группе генодерматозов, для которых характерен гиперкератоз на ладонях и подошвах без перехода на другие участки кожи.
Похожие темы научных работ по клинической медицине , автор научной работы — Пашинян Альбина Гургеновна, Ильенко Л.И., Акопян А.Н.
CLINICAL CASE OF PALMOPLANTAR KERATODERMA TYPE UNNA-THOST
Palmar-plantar keratoderma is a heterogeneous group of hereditary dermatosis , which can be independent diseases or combined with various congenital malformations, most of ectodermal origin. Approaches to differential diagnosis and clinical features of each form of this pathology are described. Keratoderma, Unna Toast genodermatosis belongs to the group, which is characterized by hyperkeratosis on the palms and soles without migration to other skin areas.
Текст научной работы на тему «Клинический случай ладонно-подошвенной кератодермии Унны-Тоста»
© КОЛЛЕКТИВ АВТОРОВ, 2017 УДК 616.5-003.871-055.5/.7
Пашинян А.Г., Ильенко Л.И., Акопян А.Н.
КЛИНИЧЕСКИЙ СЛУЧАЙ ЛАДОННО-ПОДОШВЕННОЙ КЕРАТОДЕРМИИ УННЫ-ТОСТА
ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, 117997, г. Москва, Россия
Ладонно-подошвенные кератодермии - гетерогенная группа наследственных дерматозов, которые могут быть самостоятельными заболеваниями или сочетаться с различными врожденными пороками развития, чаще всего эктодермального происхождения. Рассматривается дифференциальная диагностика, отмечаются характерные клинические признаки для каждой формы ладонно-подо-швенной кератодермии. Кератодермию Унны-Тоста относят к группе генодерматозов, для которых характерен гиперкератоз на ладонях и подошвах без перехода на другие участки кожи.
Ключевые слова: наследственные дерматозы; ладонно-подошвенные кератодермии; диффузные; очаговые формы.
Pashinyan A.G., Ilienko L.I., Akopyan A.N.
CLINICAL CASE OF PALMOPLANTAR KERATODERMA TYPE UNNA-THOST
N.I. Pirogov Russian National Research Medical University, Moscow, 117997, Russian Federation
Palmar-plantar keratoderma is a heterogeneous group of hereditary dermatosis, which can be independent diseases or combined with various congenital malformations, most of ectodermal origin. Approaches to differential diagnosis and clinicalfeatures of each form of this pathology are described. Keratoderma, Unna Toast genodermatosis belongs to the group, which is characterized by hyperkeratosis on the palms and soles without migration to other skin areas.
Keywords: hereditary dermatosis; palmar-plantar keratoderma; diffuse; focal forms.
For citation: Pashinyan A.G., Ilienko L.I., Akopyan A.N. Clinical case of palmoplantar keratoderma type Unna-Thost. Russian
Journal of Skin and Venereal Diseases (Rossiyskii Zhurnal Kozhnykh i Venericheskikh Boleznei). 2017; 20(3): 143-145.
Acknowledgments. The study had no sponsorship.
Conflict of interest. The authors declare no conflict of interest.
Received 07 Febr 2017
Accepted 26 April 2017
КЛИНИКА, ДИАГНОСТИКА И ЛЕЧЕНИЕ ДЕРМАТОЗОВ
Ладонно-подошвенные кератодермии относятся к группе заболеваний с усиленным ороговением, увеличением продукции кератиноцитов при уменьшении их нормальной десквамации [1].
Мутации в генах, кодирующих кератин 1-го и 6-го типов, ответственных за выработку кератинов и белков клеточной оболочки, могут явиться причиной развития наследственных ладонно-подошвенных кератодермий [2, 3].
Information about author:
Наследственные кератодермии могут быть как самостоятельными заболеваниями, так и сочетаться с разнообразными врожденными пороками развития, чаще всего эктодермального происхождения. Для них характерны различные типы генетического наследования (по аутосомно-доминант-ному либо по аутосомно-рецессивному типу) и многообразные клинические и морфологические особенности 4.
Помимо наследственных существует группа приобретенных кератодермий: при псориазе, красном плоском лишае, болезни Девержи, синдромах Хакстхаузена, Сезари, экземе, дерматомикозах и ряде других заболеваний, а также при паранеопластическом синдроме 7.
Ладонно-подошвенная кератодермия в зависимости от клинических проявлений подразделяется на диффузную (кератодермия Унны-Тоста, Меледа, Папийона-Лефевра, эпидермолитическая и др.) 10 и очаговую (кератодермия Сименса, линейная Фукса, Бушке-Фишера-Брауэра, краевая ладоней Рамос-и-Сильвы, акрокератоэластоидоз Косты и др.) 15.
При иммуногистохимическом и электронно-микроскопическом исследованиях биоптата выявляется нарушение строения тонофибриллярного аппарата клеток, приводя-
Рис. 1. Очаги гиперкератоза у больного 7 мес. а - на подошвах; б - на ладонях.
щее к изменению механизмов регулирования кератиниза-ции, усилению клеточной пролиферации и образованию избыточного неполноценного рогового слоя [4].
При ладонно-подошвенной кератодермии Грейтера, наследуемой по аутосомно-доминантному типу, первые признаки заболевания начинаются в возрасте от 1 года до 8 лет. Кератоз ладоней и подошв выражен незначительно, выявляется переход на тыльную поверхность ладоней и подошв, на кожу над ахилловым сухожилием. Возможно появление эритематозно-сквамозных высыпаний на разгибательных поверхностях коленных и локтевых суставов. Еще одной клинической особенностью являются нередкие случаи самопроизвольного излечения в возрасте после 40-50 лет, что не встречается при других типах диффузных ладонно-подошвенных кератодермий.
Кератодермия Меледа, передающаяся аутосомно-ре-цессивно, впервые описана среди кровных родственников населения острова Меледа. Начальные проявления болезни возникают в детском возрасте в виде стойкой эритемы с шелушением ладоней и подошв. В дальнейшем ороговение кожи усиливается и к 15-20 годам на ладонях и подошвах отмечаются массивные роговые наслоения желто-коричневого цвета, эритема сохраняется лишь в виде фиолетово-лилового ободка шириной несколько миллиметров по периферии очага. Для этой формы характерен переход кератоза с ладонно-подошвенных поверхностей на тыльную поверхность кистей, стоп, на кожу локтевых, коленных суставов с образованием болезненных глубоких трещин, особенно на пятках. Патоморфологическая картина
характеризуется гиперкератозом, иногда акантозом, в дерме - воспалительным лимфогистиоцитарным инфильтратом. Выражен локальный гипергидроз с черными точками выводных протоков потовых желез. Возможно сочетание с атопическим дерматитом. Могут отмечаться изменения на электроэнцефалограмме, умственная отсталость, синдактилия, складчатый язык, готическое нёбо [10, 21].
Кератодермия Папийона-Лефевра - наследственная диффузная форма болезни, передающаяся по аутосом-но-рецессивному типу и сочетающаяся с пародонтозом и пиогенными (гнойными) инфекциями кожи и десен. У больных отмечают снижение функции щитовидной и поджелудочной железы, нарушение функциональной активности лейкоцитов, снижение фагоцитарной активности нейтрофилов и чувствительности Т- и В-лимфоцитов к митогенам. Клинические проявления возникают в возрасте от 1 года до 5 лет (чаще на 2-3-м году жизни) в виде эритемы ладоней и подошв, покрывающихся роговыми наслоениями, интенсивность которых постепенно усиливается. Участки кератоза нередко выходят за пределы ла-донно-подошвенных поверхностей на тыл кистей и стоп, область пяточного (ахиллова) сухожилия, коленных и локтевых суставов. Характерен локализованный гипергидроз. Гистологически выявляют гиперкератоз, нерегулярный па-ракератоз, в дерме - небольшой воспалительный инфильтрат. В клетках рогового и зернистого слоев обнаруживают липидоподобные вакуоли, нарушение структуры тонофи-брилл и кератогиалиновых гранул. Ногти нередко дистро-фичные, тусклые, ломкие, волосы не изменены. В возрасте 4-5 лет в результате персистирующего гингивита развивается прогрессирующий пародонтоз с образованием гнойных альвеолярных карманов, воспалением и дистрофией альвеолярных отростков с преждевременным кариесом и выпадением зубов, аномалий их развития. Возможны каль-цификация твердой мозговой оболочки, арахнодактилия, акроостеолиз [11, 22, 23].
Кератодермия диссеминированная Бушке-Фишера-Брауэра (кератоз точечный рассеянный Бушке-Фишера) наследуется аутосомно-доминантно. Первые симптомы появляются в возрасте 15-30 лет. На коже ладоней и подошв образуются роговые узелки полигональной формы - «жемчужины» величиной около 2-10 мм в диаметре, располагающиеся изолированно или группами. При отторжении центральных роговых масс остается кратерообразное углубление, а при проведении рукой по их поверхности возникает ощущение «терки». Потоотделение не нарушено. Гистологически выявляют гиперкератоз с паракератозом в центральной части, небольшой
Рис. 2. Диффузный гиперкератоз на ладонях у мамы больного гиперкератозом ребенка.
акантоз, в дерме - незначительный периваскулярный воспалительный инфильтрат [14, 24, 25].
Краевая кератодермия ладоней Рамос-и-Сильвы - приобретенная форма кератодермии, развивающаяся у больных со злокачественными новообразованиями внутренних органов, артритами, нарушением функции половых желез, на месте травматизации кожи [15, 26].
Лечение кератодермий включает системные ретиноиды, аевит, кератолитические мази, физиопроцедуры.
Приводим наше клиническое наблюдение случая диффузной кератодермии ладоней и подошв Унны-Тоста.
На прием обратились родители мальчика 7 мес, у которого отмечаются утолщения на ладонях и подошвах. Из анамнеза известно, что у всех родственников по материнской линии (у мамы пациента и ее родного брата, у бабушки, прадедушки) отмечаются очаги гиперкератоза на ладонях и подошвах. Два других родных брата мамы здоровы. Мама ребенка к врачу по поводу своих проявлений на ладонях и подошвах не обращалась, не лечилась.
Мальчик родился от 1-й беременности, протекавшей без особенностей. Роды срочные, самостоятельные, ребенок закричал сразу. Оценка по шкале Апгар - 8/9 баллов. Период новорожденности протекал без особенностей. Ребенок растет и развивается соответственно возрасту, аллергологический анамнез не отягощен, сопутствующих заболеваний нет. Наследственность отягощена - у матери наследственная ладонно-подошвенная кератодермия.
Объективно обнаружено: у пациента на подошвах отмечаются плотные гиперкератотические воскообразные наслоения с четкими краями без перехода на другие участки кожи. На ладонях на фоне очагов гиперкератоза - крупнопластинчатое шелушение (рис. 1). Ногтевые пластинки без патологии. При микроскопическом исследовании чешуек нити мицелия не обнаружены.
У мамы выявлены диффузное утолщение ладоней, подошв, подчёркнутый кожный рисунок, трещины, истыканность ногтевых пластинок. Волосы и зубы не изменены (рис. 2).
От обследования в генетическом центре родители ребенка отказались.
Данный случай представляет клинический интерес как редко встречающееся проявление наследственной диффузной ладонно-подошвенной кератодермии для своевременной постановки диагноза, а также для привлечения внимания широкого круга неонатологов и дерматологов к
проблеме врожденных и наследственных заболеваний, сопровождающихся поражением кожи. Кроме того, установление генной природы болезни имеет существенное значение с позиций медико-генетического консультирования семьи по прогнозу здорового потомства.
Финансирование. Исследование не имело спонсорской поддержки. Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
1. Itin P.H., Fistarol S.K. Palmoplantar keratodermas. Clin. Dermatol. 2005; 23(1): 15-22.
2. Allan C.M., Procaccia S., Tran D., Tu Y., Barnes R.H., Larsson M., et al. Palmoplantar keratoderma in slurp2-deficient mice. J. Invest. Dermatol. 2016; 136(2): 436-43. doi: 10.1016/jjid.2015.11.003
3. Nomura T., Mizuno O., Miyauchi T., Suzuki S., Shinkuma S., Hata H., et all Striate palmoplantar keratoderma: Report of a novel DSG1 mutation and atypical clinical manifestations. J. Dermatol. Sci. 2015; 80(3): 223-5. doi: 10.1016/jjdermsci.2015.10.004.
4. Stypczynska E., Placek W., Zegarska B., Czajkowski R. Keratinization disorders and genetic aspects in palmar and plantar keratodermas. Acta Der-matovenerol Croat. 2016; 24(2): 116-23.
5. Sakiyama T., Kubo A. Hereditary palmoplantar keratoderma "clinical and genetic differential diagnosis". J. Dermatol. 2016; 43(3): 264-74. doi: 10.1111/1346-8138.13219.
6. Sehgal V.N., Aggarwal A,. Syed N.H., Rasool F., Verma P., Sharma S. Palmoplantar keratoderma as a variant oflichen planus. Skinmed. 2016; 14(1): 56-60.
7. Kim J., Foster R., Lam M., Kumarasinghe S.P. Mycosis fungoides: an important differential diagnosis for acquired palmoplantar keratoderma. Aus-tralas J. Dermatol 2015; 56(1): 49-51. doi: 10.1111/ajd.12155.
8. Hersle K., Mobacken H. Hyperkeratotic dermatitis of the palms. Br. J. Der-matol. 1982; 107(2): 195-201.
9. Küster W., Becker A. Indication for the identity of palmoplantar keratoderma type Unna-Thost with type Vorner. Thost's family revisited 110 years later. Acta Derm. Venereol. 1992; 72(2): 120-2.
10. Perez C., Khachemoune A. Mal de Meleda: a focused review. Am. J. Clin. Dermatol. 2016; 17(1): 63-70. doi: 10.1007/s40257-015-0157-1.
11. Jijin M.J., Jaishankar H.P., Narayaran V.S., Rangaswamy K., Puthaswamy K.A. Papillon-Lefevre syndrome in an adolescent female: a case study. Clin. Diagn. Res. 2015; 9(5): 23-5. doi: 10.7860/JCDR/2015/12780.5921.
12. Bragg J., Rizzo C., Mengden S. Striate palmoplantar keratoderma (Brunau-er-Fohs-Siemens syndrome). Dermatol. Online J. 2008; 14(5): 26.
13. Schiller S., Seebode C., Hennies H., Giehl K., Emmert S. Palmoplantar keratoderma (PPK): acquired and genetic causes of a not so rare disease. J. Dtsch. Dermatol Ges. 2014; 12(9): 781-8. doi: 10.1111/ddg.12418.
14. Antonio J.R., Oliveira G.B., Rossi N.C., Pires L.G. Exuberant clinical picture of Buschke-Fischer-Brauer palmoplantar keratoderma in bedridden patient. An Bras. Dermatol. 2014; 89(5): 819-21.
15. Ramos-E-Silva M., Carvalho J.C., Carneiro S.C. Cutaneous paraneoplasia. Clin. Dermatol. 2011; 29(5): 541-7.
16. Hu W., Cook T.F., Vicki G.J., Glaser D.A. Acrokeratoelastoidosis. Pediatr. Dermatol. 2002; 19(4): 320-2.
20. Sehgal V.N., Sardana K., Sharma S., Raut D. Hereditary palmoplantar (epi-dermolytic) keratoderma: illustration through a familial report. Skinmed. 2004; 3(6): 323-30.
21. Bakija-Konsuo A. Mal de Meleda - through history and today. Acta Der-matovenerol. Croat. 2014; 22(2): 79-84.
22. Iqtadar S., Mumtaz S.U., Abaidullah S. Papillon-Lefevre syndrome with palmoplantar keratoderma and periodontitis, a rare cause of pyrexia of unknown origin: a case report. J. Med. Case Rep. 2015; 9: 288. doi: 10.1186/ s13256-015-0773-7.
23. Janjua S.A., Khachemoune A. Papillon-Lefevre syndrome: case report and review of the literature. Dermatol. Online J. 2004; 10(1): 13.
24. Mallo S., Bernal A.I., Fernández-Canedo M.I., González-Hermoso C., de Troya-Martín M. Autosomal dominant punctate palmoplantar keratoderma. Actas Dermosifiliogr. 2006; 97(2): 136-8.
Читайте также: